Computational Analysis of the Energetic Stability of High-Entropy Structures of a Prototypical Lanthanide-Based Metal–Organic Framework
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
High-entropy materials are characterized by their complex compositions, typically comprising five or more elements in near-equiatomic proportions. Applying this concept to metal ions in metal–organic frameworks (MOFs) has paved the way for exploring a new class of high-entropy MOFs. While the compositional strategy of high-entropy materials leverages configurational entropy to aid thermodynamic stability, it also poses significant analytical challenges due to the vast compositional landscape and diverse phases that these materials can adopt. We present a computational study of several complexities associated with selecting potential high-entropy versions of a prototype lanthanide-based MOF. We compute the energetics of metal mixing of these heterometallic MOFs using density functional theory (DFT) and machine learning interatomic potential (MLIP) methods. The use of MLIP methods allows a systematic exploration of the convex hull of thermodynamically stable MOF structures containing up to 5 distinct metals.
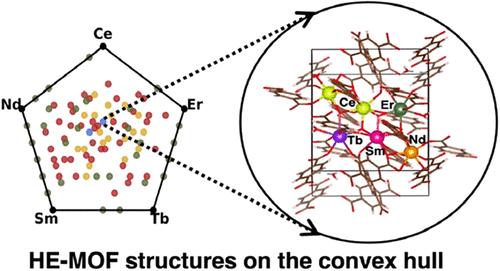
典型镧系金属-有机骨架高熵结构能量稳定性的计算分析
高熵材料以其复杂的组成为特征,通常由五种或更多元素以接近等原子的比例组成。将这一概念应用于金属有机骨架(mof)中的金属离子,为探索一类新的高熵mof铺平了道路。虽然高熵材料的组成策略利用构型熵来帮助热力学稳定性,但由于这些材料可以采用巨大的组成景观和不同的阶段,它也带来了重大的分析挑战。我们提出了与选择基于镧系元素的MOF原型的潜在高熵版本相关的几个复杂性的计算研究。我们利用密度泛函理论(DFT)和机器学习原子间势(MLIP)方法计算了这些异质金属mof的金属混合热力学。使用MLIP方法可以系统地探索包含多达5种不同金属的热力学稳定MOF结构的凸壳。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


