Esther Rubio-Portillo, Rebeca Arias-Real, Esther Rodríguez-Pérez, Lluis Bañeras, Josefa Antón, Asunción de Los Ríos
{"title":"Short-term virus-host interactions and functional dynamics in recently deglaciated Antarctic tundra soils.","authors":"Esther Rubio-Portillo, Rebeca Arias-Real, Esther Rodríguez-Pérez, Lluis Bañeras, Josefa Antón, Asunción de Los Ríos","doi":"10.1093/ismeco/ycaf157","DOIUrl":null,"url":null,"abstract":"<p><p>Long-term chronosequence studies have shown that, as glaciers retreat, newly exposed soils become colonized through primary succession. To determine the key drivers of this process and their vulnerability to climate change, the short-term responses of these pioneering microbial communities also need to be elucidated. Here, we investigated how the taxonomic and functional structure of microbial communities, including viruses, changed over a 7-year period in an Antarctic glacier forefield. Using metagenomics and metatranscriptomics we assessed the influence of both abiotic and biotic factors on these communities. Our results revealed a highly heterogeneous bacteria-dominated microbial community, with <i>Pseudomonas</i> as the most abundant genus, followed by <i>Lysobacter</i>, <i>Devosia</i>, <i>Cellulomonas</i>, and <i>Brevundimonas</i>. This community exhibited the capacity for aerobic anoxygenic phototrophy, carbon and nitrogen fixation, and sulfur cycling, processes vital for survival in nutrient-poor environments. 52 high-quality metagenome-assembled genomes (MAGs) were recovered, representing both transient and cosmopolitan taxa, some of which were able to rapidly respond to environmental changes. A diverse and highly dynamic collection of lytic and temperate viruses was identified across all samples, with high clonal viral genomes typically detected in only one of the eight samples analyzed. Metatranscriptomic analyses confirmed the activity of lytic viruses, while prophage genomes featured much lower expression levels. Prophages appeared to influence host fitness through the expression of genes encoding membrane transporters. Additionally, the abundance of genes linked to antimicrobial compound synthesis and resistance, along with antiphage defense systems, highlights the importance of biotic interactions in driving microbial community succession and shaping short-term responses to environmental fluctuations.</p>","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":"5 1","pages":"ycaf157"},"PeriodicalIF":6.1000,"publicationDate":"2025-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12507030/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/ismeco/ycaf157","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
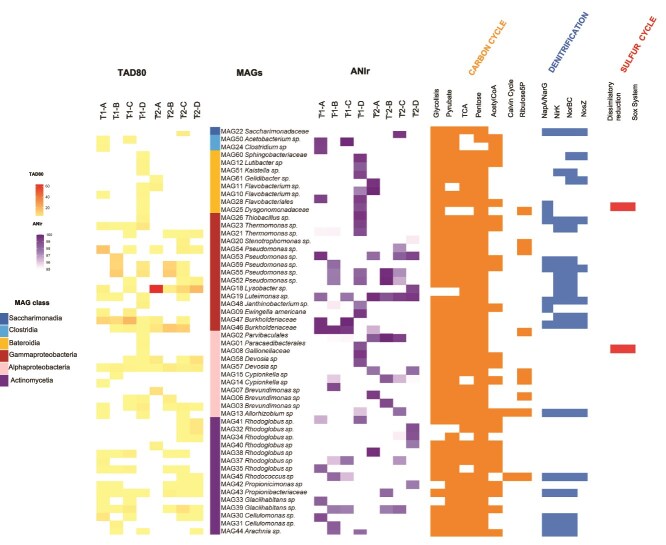
Long-term chronosequence studies have shown that, as glaciers retreat, newly exposed soils become colonized through primary succession. To determine the key drivers of this process and their vulnerability to climate change, the short-term responses of these pioneering microbial communities also need to be elucidated. Here, we investigated how the taxonomic and functional structure of microbial communities, including viruses, changed over a 7-year period in an Antarctic glacier forefield. Using metagenomics and metatranscriptomics we assessed the influence of both abiotic and biotic factors on these communities. Our results revealed a highly heterogeneous bacteria-dominated microbial community, with Pseudomonas as the most abundant genus, followed by Lysobacter, Devosia, Cellulomonas, and Brevundimonas. This community exhibited the capacity for aerobic anoxygenic phototrophy, carbon and nitrogen fixation, and sulfur cycling, processes vital for survival in nutrient-poor environments. 52 high-quality metagenome-assembled genomes (MAGs) were recovered, representing both transient and cosmopolitan taxa, some of which were able to rapidly respond to environmental changes. A diverse and highly dynamic collection of lytic and temperate viruses was identified across all samples, with high clonal viral genomes typically detected in only one of the eight samples analyzed. Metatranscriptomic analyses confirmed the activity of lytic viruses, while prophage genomes featured much lower expression levels. Prophages appeared to influence host fitness through the expression of genes encoding membrane transporters. Additionally, the abundance of genes linked to antimicrobial compound synthesis and resistance, along with antiphage defense systems, highlights the importance of biotic interactions in driving microbial community succession and shaping short-term responses to environmental fluctuations.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


