Study of the pericyclic [2π + 4π] nature of a set of cheletropic reactions: analysis of the electronic reaction mechanism through bond reactivity descriptors and the electronic bonding structure
{"title":"Study of the pericyclic [2π + 4π] nature of a set of cheletropic reactions: analysis of the electronic reaction mechanism through bond reactivity descriptors and the electronic bonding structure","authors":"Jesús Sánchez-Márquez, Alejandro Morales-Bayuelo","doi":"10.1007/s00894-025-06510-9","DOIUrl":null,"url":null,"abstract":"<p>Cheletropic reactions are a class of pericyclic transformations with significant importance in synthetic organic chemistry. Traditionally explained through orbital symmetry considerations under the Woodward–Hoffmann rules, these reactions are often modeled using wavefunction-based methods. However, this study adopts an electron-density-centered approach, with the aim of providing a detailed description and explanation of the studied reactions using reactivity descriptors derived from Density Functional Theory. By analyzing electronic bonding structures, natural bond orbitals, and bond reactivity indices, we aim to offer a more detailed understanding of the stereoelectronic factors governing the [2π + 4π] nature of these processes. This framework enables the identification of subtle features such as charge delocalization and bond reorganization at the transition states, contributing to a refined theoretical model for pericyclic reactivity. The methodology may also support the rational design of new stereoselective reactions based on local electronic properties.</p><p>All quantum chemical calculations were performed using Gaussian 16. Geometries and vibrational frequencies were calculated at the B3LYP/6-31G(d,p) level to identify stationary points. IRC calculations with a stepsize of 0.1 amu<sup>1</sup>/<sup>2</sup>·bohr confirmed the connectivity of transition states to reactants and products. Single-point energy refinements were carried out using the MPWB1K functional and the 6-311G(d,p) basis set. To obtain accurate electron densities, further calculations were performed at the CAM-B3LYP/aug-cc-pVTZ level. Natural bond orbital (NBO) analysis (v3.0) was used to evaluate donor–acceptor interactions via second-order perturbation theory. The Quantum Theory of Atoms in Molecules (QTAIM) was applied using AIMAll to locate and analyze bond critical points (BCPs). Non-covalent interactions (NCI) were examined with Multiwfn 3.8 to identify regions of weak interactions. Bond reactivity descriptors were computed using UCA-FUKUI v.2.1, a code based on conceptual-DFT and the electronegativity equalization principle. This method evaluates local reactivity without relying on atomic Fukui function partitioning, using bonding orbitals as the basis for descriptor calculation.</p>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06510-9","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
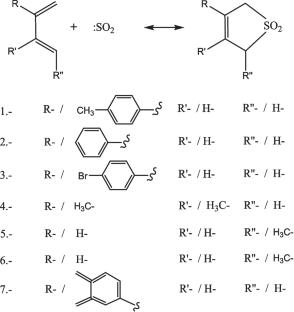
Cheletropic reactions are a class of pericyclic transformations with significant importance in synthetic organic chemistry. Traditionally explained through orbital symmetry considerations under the Woodward–Hoffmann rules, these reactions are often modeled using wavefunction-based methods. However, this study adopts an electron-density-centered approach, with the aim of providing a detailed description and explanation of the studied reactions using reactivity descriptors derived from Density Functional Theory. By analyzing electronic bonding structures, natural bond orbitals, and bond reactivity indices, we aim to offer a more detailed understanding of the stereoelectronic factors governing the [2π + 4π] nature of these processes. This framework enables the identification of subtle features such as charge delocalization and bond reorganization at the transition states, contributing to a refined theoretical model for pericyclic reactivity. The methodology may also support the rational design of new stereoselective reactions based on local electronic properties.
All quantum chemical calculations were performed using Gaussian 16. Geometries and vibrational frequencies were calculated at the B3LYP/6-31G(d,p) level to identify stationary points. IRC calculations with a stepsize of 0.1 amu1/2·bohr confirmed the connectivity of transition states to reactants and products. Single-point energy refinements were carried out using the MPWB1K functional and the 6-311G(d,p) basis set. To obtain accurate electron densities, further calculations were performed at the CAM-B3LYP/aug-cc-pVTZ level. Natural bond orbital (NBO) analysis (v3.0) was used to evaluate donor–acceptor interactions via second-order perturbation theory. The Quantum Theory of Atoms in Molecules (QTAIM) was applied using AIMAll to locate and analyze bond critical points (BCPs). Non-covalent interactions (NCI) were examined with Multiwfn 3.8 to identify regions of weak interactions. Bond reactivity descriptors were computed using UCA-FUKUI v.2.1, a code based on conceptual-DFT and the electronegativity equalization principle. This method evaluates local reactivity without relying on atomic Fukui function partitioning, using bonding orbitals as the basis for descriptor calculation.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


