Nicholas Jones, Matthew Nguyen, Dao Le, Ramy Hanna
{"title":"Novel OFD1 Mutation Results in Unusually Early-Onset Polycystic Kidney Disease.","authors":"Nicholas Jones, Matthew Nguyen, Dao Le, Ramy Hanna","doi":"10.1159/000547878","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Mutations of the OFD1 gene cause oral-facial-digital syndrome type 1 (OFD 1) and variations of the related ciliopathies Joubert syndrome and primary ciliary dyskinesia. OFD 1 manifests with skeletal, CNS, and renal abnormalities with prevalence estimated between 1 in 50,000 and 1 in 250,000. Cilia help regulate responses to mechanical forces in the renal tubules, and polycystic kidney disease (PKD) resulting from defective cilia is the primary determinant of morbidity in OFD 1. PKD associated with OFD 1 is rare before adulthood but may increase markedly with age, progressing to end-stage renal disease (ESRD) in 80% of cases.</p><p><strong>Case presentation: </strong>We report an 18-year-old female with congenital ciliopathy presenting with declining renal function and an increase of serum creatinine to 1.7 mg/dL. A 24-h urine collection yielded 0.8 g of creatinine and 500 mg of total protein. Imaging was conducted and genetic studies were repeated as her early childhood results were not available. MRI revealed numerous bilateral renal cysts consistent with progression of ciliopathy-associated PKD. Genetic testing confirmed the presence of a novel c.1332del frameshift mutation in the OFD1 gene, prematurely truncating the OFD1 protein. Modifications to diet and hydration to preserve renal function and delay progression to ESRD were initiated.</p><p><strong>Conclusion: </strong>This case of unusually early renal decline highlights the challenge of treating OFD 1. There are no currently approved medications and management consists of supportive measures to delay progression. Further research to better characterize and identify treatments for OFD 1 and related ciliopathies is warranted.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"15 1","pages":"216-223"},"PeriodicalIF":0.9000,"publicationDate":"2025-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12503729/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000547878","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Mutations of the OFD1 gene cause oral-facial-digital syndrome type 1 (OFD 1) and variations of the related ciliopathies Joubert syndrome and primary ciliary dyskinesia. OFD 1 manifests with skeletal, CNS, and renal abnormalities with prevalence estimated between 1 in 50,000 and 1 in 250,000. Cilia help regulate responses to mechanical forces in the renal tubules, and polycystic kidney disease (PKD) resulting from defective cilia is the primary determinant of morbidity in OFD 1. PKD associated with OFD 1 is rare before adulthood but may increase markedly with age, progressing to end-stage renal disease (ESRD) in 80% of cases.
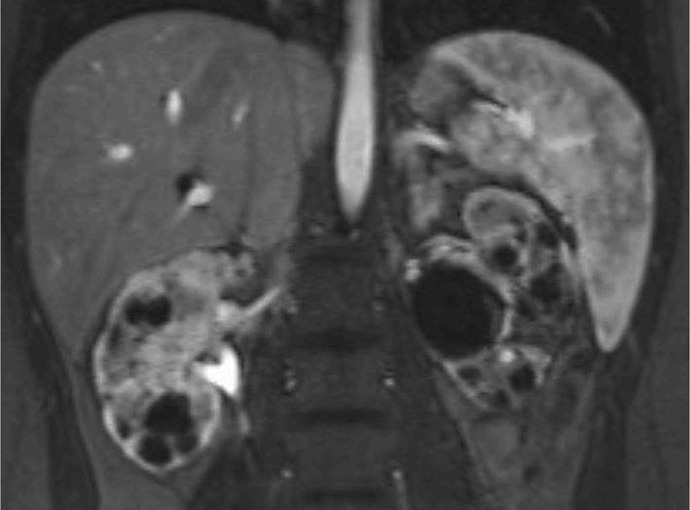
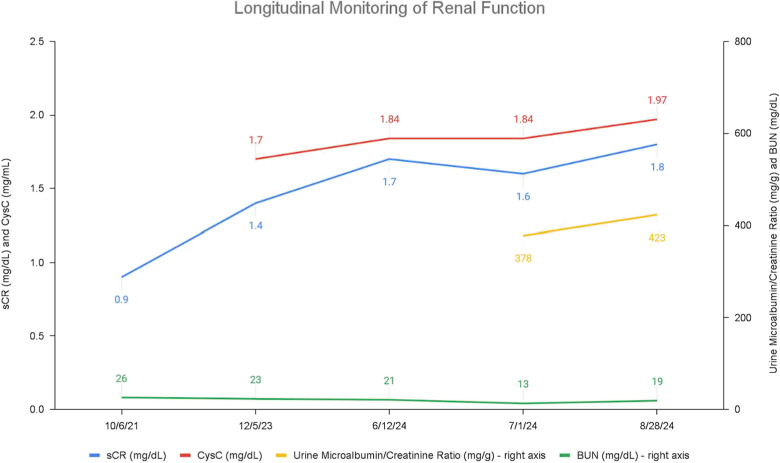
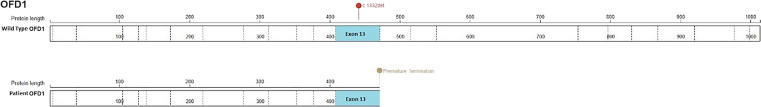
Case presentation: We report an 18-year-old female with congenital ciliopathy presenting with declining renal function and an increase of serum creatinine to 1.7 mg/dL. A 24-h urine collection yielded 0.8 g of creatinine and 500 mg of total protein. Imaging was conducted and genetic studies were repeated as her early childhood results were not available. MRI revealed numerous bilateral renal cysts consistent with progression of ciliopathy-associated PKD. Genetic testing confirmed the presence of a novel c.1332del frameshift mutation in the OFD1 gene, prematurely truncating the OFD1 protein. Modifications to diet and hydration to preserve renal function and delay progression to ESRD were initiated.
Conclusion: This case of unusually early renal decline highlights the challenge of treating OFD 1. There are no currently approved medications and management consists of supportive measures to delay progression. Further research to better characterize and identify treatments for OFD 1 and related ciliopathies is warranted.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


