Akshat Shirish Zalte, Hao-Wei Pang, Anna C Doner, William H Green
{"title":"RIGR: Resonance-Invariant Graph Representation for Molecular Property Prediction.","authors":"Akshat Shirish Zalte, Hao-Wei Pang, Anna C Doner, William H Green","doi":"10.1021/acs.jcim.5c00495","DOIUrl":null,"url":null,"abstract":"<p><p>Many successful machine learning models for molecular property prediction rely on Lewis structure representations, commonly encoded as SMILES strings. However, a key limitation arises with molecules exhibiting resonance, where multiple valid Lewis structures represent the same species. This causes inconsistent predictions for the same molecule based on the chosen resonance form in common property prediction frameworks such as Chemprop, which implements a directed message-passing neural network (D-MPNN) architecture on the input molecular graph. To address this issue of resonance variance, we introduce the resonance-invariant graph representation (RIGR) of molecules that ensures, by construction, that all resonance structures are mapped to a single representation, eliminating the need to choose from or generate multiple resonance structures. Implemented with the D-MPNN architecture, RIGR is evaluated on a large data set with resonance-exhibiting radicals and closed-shell molecules, comparing it against the Chemprop featurizer. Using 60% fewer features, RIGR demonstrates comparable or superior prediction performance. Alternative approaches, such as data augmentation with resonance forms, are assessed, and their limitations are explored. Available open-source as an optional featurization scheme in Chemprop, RIGR is benchmarked across a wide range of property prediction tasks, showcasing its potential as a general graph featurizer beyond resonance handling.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2025-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.5c00495","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
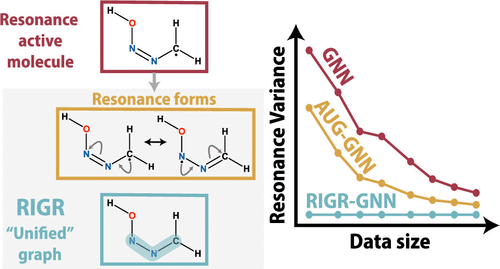
Many successful machine learning models for molecular property prediction rely on Lewis structure representations, commonly encoded as SMILES strings. However, a key limitation arises with molecules exhibiting resonance, where multiple valid Lewis structures represent the same species. This causes inconsistent predictions for the same molecule based on the chosen resonance form in common property prediction frameworks such as Chemprop, which implements a directed message-passing neural network (D-MPNN) architecture on the input molecular graph. To address this issue of resonance variance, we introduce the resonance-invariant graph representation (RIGR) of molecules that ensures, by construction, that all resonance structures are mapped to a single representation, eliminating the need to choose from or generate multiple resonance structures. Implemented with the D-MPNN architecture, RIGR is evaluated on a large data set with resonance-exhibiting radicals and closed-shell molecules, comparing it against the Chemprop featurizer. Using 60% fewer features, RIGR demonstrates comparable or superior prediction performance. Alternative approaches, such as data augmentation with resonance forms, are assessed, and their limitations are explored. Available open-source as an optional featurization scheme in Chemprop, RIGR is benchmarked across a wide range of property prediction tasks, showcasing its potential as a general graph featurizer beyond resonance handling.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


