Xiucheng Jiang, Lan Shi, Mei Zhao, Cui Chen, Tao Tang, Simeng Ji, Bingbing Lv, Lihua Jia, Shuhan Duan, Jinyue Ma, Jiyu Pang, Bo Mu, Yongsheng Zhao, Junbao Yang
{"title":"Next-generation sequencing of mitochondrial DNA reveals pathogenic variants and protective haplogroup D4 in esophageal cancer.","authors":"Xiucheng Jiang, Lan Shi, Mei Zhao, Cui Chen, Tao Tang, Simeng Ji, Bingbing Lv, Lihua Jia, Shuhan Duan, Jinyue Ma, Jiyu Pang, Bo Mu, Yongsheng Zhao, Junbao Yang","doi":"10.3389/fgene.2025.1643229","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>The germline variations in the mitochondrial genome of esophageal cancer (EC) remain uncertain. Our study aimed to explore the distribution and pathogenicity of mitochondrial genome variations in EC, as well as to identify haplogroups associated with the development of EC.</p><p><strong>Methods: </strong>We performed next-generation sequencing of the mitochondrial genomes from peripheral blood samples of 146 EC patients and 120 healthy controls. Variant annotation was performed using MitoMap, while pathogenicity prediction was conducted through tools such as MitoTip, SIFT, and PolyPhen2. Moreover, haplogroup classification was carried out using the Haplogrep3 platform.</p><p><strong>Results: </strong>A total of 1299 mitochondrial variants were identified among 146 EC patients, including 171 novel (previously unreported) mutations. Compared with the healthy control group, the EC cohort exhibited a higher frequency of variants in genes such as ND2, COX1, COX2, 12S rRNA, and 16S rRNA. Three tRNA mutations (7496_T>C, 5771_A>G, and 5613_T>A) were predicted to be potentially pathogenic. Within the protein-coding regions, 14 variants were classified as deleterious based on predictions from 13 independent bioinformatic algorithms. Notably, mitochondrial haplogroup D4 was significantly associated with a decreased risk of developing EC. Furthermore, several mtDNA single-nucleotide polymorphisms (SNPs), including 302_A>AC, 1824_T>C, 1842_A>G, 3010_G>A, 8414_C>T, and 14668_C>T, showed significant associations with EC susceptibility.</p><p><strong>Conclusion: </strong>We found that the number of variations in multiple regions of the mitochondrial genome in the EC population was higher than that in the control group. Additionally, several potentially pathogenic variants were identified, and haplogroup D4 was suggested as a potentially protective haplogroup against the development of EC.</p>","PeriodicalId":12750,"journal":{"name":"Frontiers in Genetics","volume":"16 ","pages":"1643229"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12497592/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.3389/fgene.2025.1643229","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: The germline variations in the mitochondrial genome of esophageal cancer (EC) remain uncertain. Our study aimed to explore the distribution and pathogenicity of mitochondrial genome variations in EC, as well as to identify haplogroups associated with the development of EC.
Methods: We performed next-generation sequencing of the mitochondrial genomes from peripheral blood samples of 146 EC patients and 120 healthy controls. Variant annotation was performed using MitoMap, while pathogenicity prediction was conducted through tools such as MitoTip, SIFT, and PolyPhen2. Moreover, haplogroup classification was carried out using the Haplogrep3 platform.
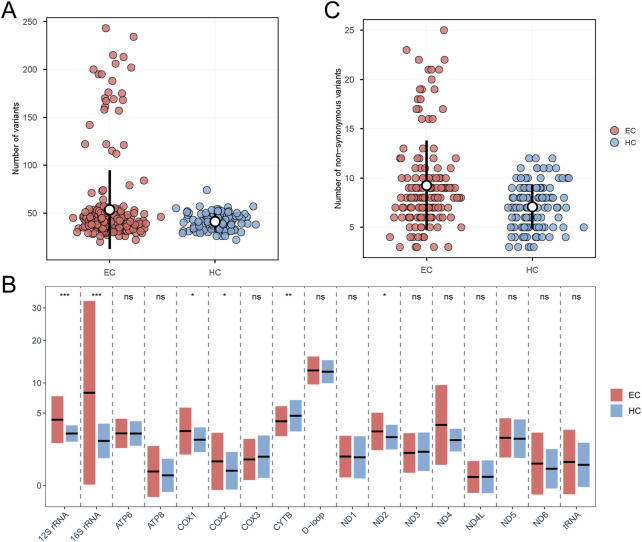
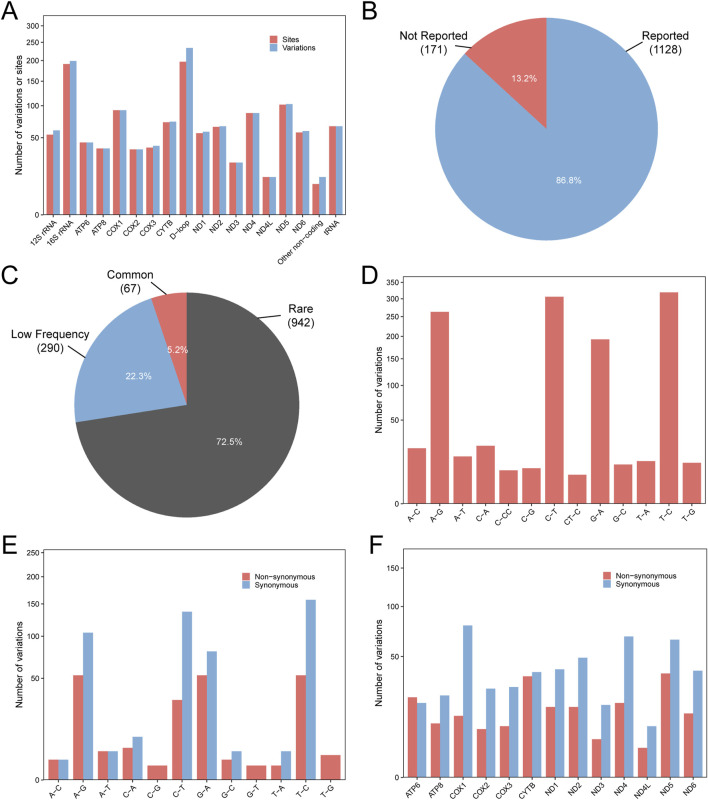
Results: A total of 1299 mitochondrial variants were identified among 146 EC patients, including 171 novel (previously unreported) mutations. Compared with the healthy control group, the EC cohort exhibited a higher frequency of variants in genes such as ND2, COX1, COX2, 12S rRNA, and 16S rRNA. Three tRNA mutations (7496_T>C, 5771_A>G, and 5613_T>A) were predicted to be potentially pathogenic. Within the protein-coding regions, 14 variants were classified as deleterious based on predictions from 13 independent bioinformatic algorithms. Notably, mitochondrial haplogroup D4 was significantly associated with a decreased risk of developing EC. Furthermore, several mtDNA single-nucleotide polymorphisms (SNPs), including 302_A>AC, 1824_T>C, 1842_A>G, 3010_G>A, 8414_C>T, and 14668_C>T, showed significant associations with EC susceptibility.
Conclusion: We found that the number of variations in multiple regions of the mitochondrial genome in the EC population was higher than that in the control group. Additionally, several potentially pathogenic variants were identified, and haplogroup D4 was suggested as a potentially protective haplogroup against the development of EC.
Frontiers in GeneticsBiochemistry, Genetics and Molecular Biology-Molecular Medicine
CiteScore
5.50
自引率
8.10%
发文量
3491
审稿时长
14 weeks
期刊介绍:
Frontiers in Genetics publishes rigorously peer-reviewed research on genes and genomes relating to all the domains of life, from humans to plants to livestock and other model organisms. Led by an outstanding Editorial Board of the world’s leading experts, this multidisciplinary, open-access journal is at the forefront of communicating cutting-edge research to researchers, academics, clinicians, policy makers and the public.
The study of inheritance and the impact of the genome on various biological processes is well documented. However, the majority of discoveries are still to come. A new era is seeing major developments in the function and variability of the genome, the use of genetic and genomic tools and the analysis of the genetic basis of various biological phenomena.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


