Colin Bousige*, , , Anouar-Akacha Delenda, , , Abdul-Rahman Allouche, , and , Pierre Mignon,
{"title":"A Portable Data Set for Borophene Growth Modeling with Reactive Neural Network Potentials","authors":"Colin Bousige*, , , Anouar-Akacha Delenda, , , Abdul-Rahman Allouche, , and , Pierre Mignon, ","doi":"10.1021/acs.jpcc.5c04912","DOIUrl":null,"url":null,"abstract":"<p >In this study, we develop and validate machine learned interaction potentials (MLIPs) for reactive simulation of borophene on metal substrates. A versatile training data set is constructed to accurately represent both extended and reactive borophene structures. It should be portable to train any MLIP architecture. Indeed, three generations of MLIPs, namely n2p2, DeePMD and NNMP, are trained and validated against density functional theory (DFT) calculations. Our results demonstrate the capability of the MLIPs to accurately reproduce DFT-calculated structures, energies, and forces. We finally show that it is possible to use these MLIPs to simulate the growth of borophene on a silver substrate.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 41","pages":"18760–18771"},"PeriodicalIF":3.2000,"publicationDate":"2025-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c04912","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
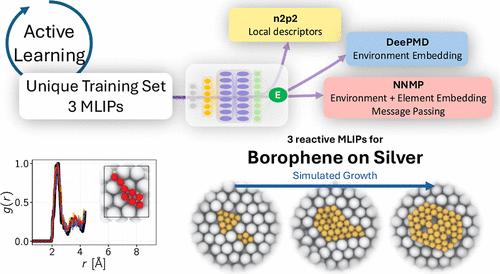
In this study, we develop and validate machine learned interaction potentials (MLIPs) for reactive simulation of borophene on metal substrates. A versatile training data set is constructed to accurately represent both extended and reactive borophene structures. It should be portable to train any MLIP architecture. Indeed, three generations of MLIPs, namely n2p2, DeePMD and NNMP, are trained and validated against density functional theory (DFT) calculations. Our results demonstrate the capability of the MLIPs to accurately reproduce DFT-calculated structures, energies, and forces. We finally show that it is possible to use these MLIPs to simulate the growth of borophene on a silver substrate.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


