{"title":"DIRseq as a method for predicting drug-interacting residues of intrinsically disordered proteins from sequences.","authors":"Matt MacAinsh, Sanbo Qin, Huan-Xiang Zhou","doi":"10.7554/eLife.107470","DOIUrl":null,"url":null,"abstract":"<p><p>Intrinsically disordered proteins (IDPs) are now well-recognized as drug targets. Identifying drug-interacting residues is valuable for both optimizing compounds and elucidating the mechanism of action. Currently, NMR chemical shift perturbation and all-atom molecular dynamics (MD) simulations are the primary tools for this purpose. Here, we present DIRseq, a fast method for predicting drug-interacting residues from the amino-acid sequence. All residues contribute to the propensity of a particular residue to be drug-interacting; the contributing factor of each residue has an amplitude that is determined by its amino-acid type and attenuates with increasing sequence distance from the particular residue. DIRseq predictions match well with drug-interacting residues identified by NMR chemical shift perturbation and other methods, including residues L<sub>22</sub>WK<sub>24</sub> and Q<sub>52</sub>WFT<sub>55</sub> in the tumor suppressor protein p53. These successes augur well for deciphering the sequence code for IDP-drug binding. DIRseq is available as a web server at https://zhougroup-uic.github.io/DIRseq/ and has many applications, such as virtual screening against IDPs and designing IDP fragments for in-depth NMR and MD studies.</p>","PeriodicalId":11640,"journal":{"name":"eLife","volume":"14 ","pages":""},"PeriodicalIF":6.4000,"publicationDate":"2025-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12503486/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"eLife","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.7554/eLife.107470","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
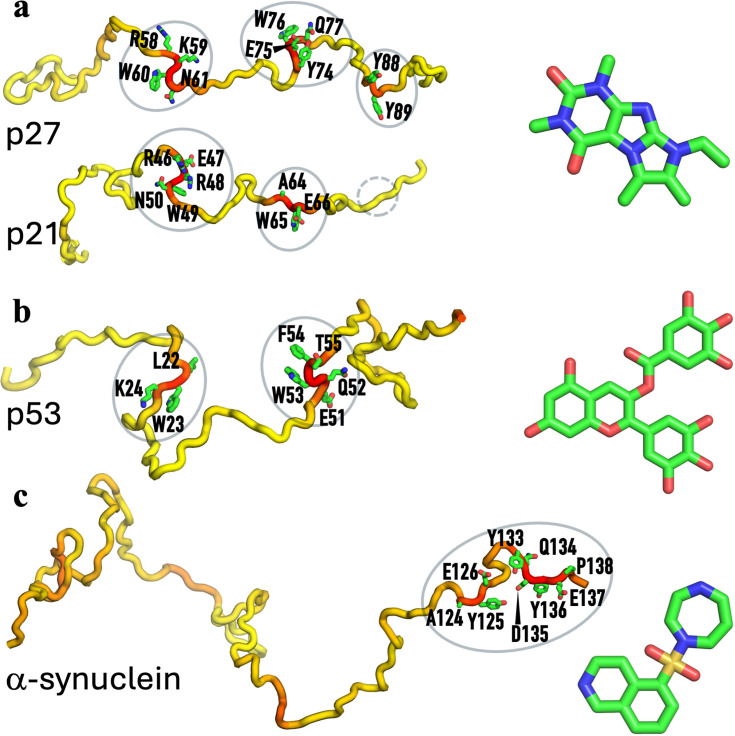
Intrinsically disordered proteins (IDPs) are now well-recognized as drug targets. Identifying drug-interacting residues is valuable for both optimizing compounds and elucidating the mechanism of action. Currently, NMR chemical shift perturbation and all-atom molecular dynamics (MD) simulations are the primary tools for this purpose. Here, we present DIRseq, a fast method for predicting drug-interacting residues from the amino-acid sequence. All residues contribute to the propensity of a particular residue to be drug-interacting; the contributing factor of each residue has an amplitude that is determined by its amino-acid type and attenuates with increasing sequence distance from the particular residue. DIRseq predictions match well with drug-interacting residues identified by NMR chemical shift perturbation and other methods, including residues L22WK24 and Q52WFT55 in the tumor suppressor protein p53. These successes augur well for deciphering the sequence code for IDP-drug binding. DIRseq is available as a web server at https://zhougroup-uic.github.io/DIRseq/ and has many applications, such as virtual screening against IDPs and designing IDP fragments for in-depth NMR and MD studies.
期刊介绍:
eLife is a distinguished, not-for-profit, peer-reviewed open access scientific journal that specializes in the fields of biomedical and life sciences. eLife is known for its selective publication process, which includes a variety of article types such as:
Research Articles: Detailed reports of original research findings.
Short Reports: Concise presentations of significant findings that do not warrant a full-length research article.
Tools and Resources: Descriptions of new tools, technologies, or resources that facilitate scientific research.
Research Advances: Brief reports on significant scientific advancements that have immediate implications for the field.
Scientific Correspondence: Short communications that comment on or provide additional information related to published articles.
Review Articles: Comprehensive overviews of a specific topic or field within the life sciences.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


