Farshud Sorourifar, Thomas Banker and Joel A. Paulson
{"title":"Adaptive subspace Bayesian optimization over molecular descriptor libraries for data-efficient chemical design","authors":"Farshud Sorourifar, Thomas Banker and Joel A. Paulson","doi":"10.1039/D5DD00188A","DOIUrl":null,"url":null,"abstract":"<p >The discovery of molecules with optimal functional properties is a central challenge across diverse fields such as energy storage, catalysis, and chemical sensing. However, molecular property optimization (MPO) remains difficult due to the combinatorial size of chemical space and the cost of acquiring property labels <em>via</em> simulations or wet-lab experiments. Bayesian optimization (BO) offers a principled framework for sample-efficient discovery in such settings, but its effectiveness depends critically on the quality of the molecular representation used to train the underlying probabilistic surrogate model. Existing approaches based on fingerprints, graphs, SMILES strings, or learned embeddings often struggle in low-data regimes due to high dimensionality or poorly structured latent spaces. Here, we introduce Molecular Descriptors with Actively Identified Subspaces (MolDAIS), a flexible molecular BO framework that adaptively identifies task-relevant subspaces within large descriptor libraries. Leveraging the sparse axis-aligned subspace (SAAS) prior introduced in recent BO literature, MolDAIS constructs parsimonious Gaussian process surrogate models that focus on task-relevant features as new data is acquired. In addition to validating this approach for descriptor-based MPO, we introduce two novel screening variants, which significantly reduce computational cost while preserving predictive accuracy and physical interpretability. We demonstrate that MolDAIS consistently outperforms state-of-the-art MPO methods across a suite of benchmark and real-world tasks, including single- and multi-objective optimization. Our results show that MolDAIS can identify near-optimal candidates from chemical libraries with over 100 000 molecules using fewer than 100 property evaluations, highlighting its promise as a practical tool for data-scarce molecular discovery.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 10","pages":" 2910-2926"},"PeriodicalIF":6.2000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/dd/d5dd00188a?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00188a","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
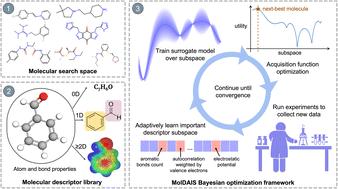
The discovery of molecules with optimal functional properties is a central challenge across diverse fields such as energy storage, catalysis, and chemical sensing. However, molecular property optimization (MPO) remains difficult due to the combinatorial size of chemical space and the cost of acquiring property labels via simulations or wet-lab experiments. Bayesian optimization (BO) offers a principled framework for sample-efficient discovery in such settings, but its effectiveness depends critically on the quality of the molecular representation used to train the underlying probabilistic surrogate model. Existing approaches based on fingerprints, graphs, SMILES strings, or learned embeddings often struggle in low-data regimes due to high dimensionality or poorly structured latent spaces. Here, we introduce Molecular Descriptors with Actively Identified Subspaces (MolDAIS), a flexible molecular BO framework that adaptively identifies task-relevant subspaces within large descriptor libraries. Leveraging the sparse axis-aligned subspace (SAAS) prior introduced in recent BO literature, MolDAIS constructs parsimonious Gaussian process surrogate models that focus on task-relevant features as new data is acquired. In addition to validating this approach for descriptor-based MPO, we introduce two novel screening variants, which significantly reduce computational cost while preserving predictive accuracy and physical interpretability. We demonstrate that MolDAIS consistently outperforms state-of-the-art MPO methods across a suite of benchmark and real-world tasks, including single- and multi-objective optimization. Our results show that MolDAIS can identify near-optimal candidates from chemical libraries with over 100 000 molecules using fewer than 100 property evaluations, highlighting its promise as a practical tool for data-scarce molecular discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


