{"title":"BC-predict: mining of signal biomarkers and production of models for early-stage breast cancer subtyping and prognosis.","authors":"Sangeetha Muthamilselvan, Natarajan Vaithilingam, Ashok Palaniappan","doi":"10.3389/fbinf.2025.1644695","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Disease heterogeneity is the hallmark of breast cancer, which is the most common female malignancy. With a disturbing increase in mortality and disease burden, there remains a need for effective early-stage theragnostic and prognostic biomarkers. In this work, we improved on BrcaDx (https://apalania.shinyapps.io/brcadx/) for cancer vs control screening and examined a cluster of adjoining learning problems in breast cancer heterogeneity: (i) identification of metastatic cancers; (ii) molecular subtyping (TNBC, HER2, or luminal); and (iii) histological subtyping (invasive ductal or invasive lobular).</p><p><strong>Methods: </strong>We analyzed the transcriptomic profiles of breast cancer patients from public-domain databases such as the TCGA using stage-encoded problem-specific statistical models of gene expression and unveiled stage-salient and progression-significant genes. Using a consensus approach, we identified potential machine learning features, and considered six model classes for each learning problem, with hyperparameter optimization on a training dataset and evaluation on a holdout test dataset. A nested approach enabled us to identify the best model class for each learning problem.</p><p><strong>Results: </strong>External validation of the best models yielded balanced accuracies of 97.42% for cancer vs normal; 88.22% for metastatic v/s non metastatic; 88.79% for ternary molecular subtyping; and ensemble accuracy of 94.23% for histological subtyping. The model for molecular subtyping was validated on a 26-sample TNBC-only out-of-distribution cohort, yielding 25 correct predictions. We performed a late integration of multi-omics datasets by validating the feature space used in each problem with miRNA profiles, methylation profiles, and commercial breast cancer panels.</p><p><strong>Discussion: </strong>Pending prospective studies, we have translated the models into BC-Predict that forks the best models developed for each problem in a unified interface and provides a complete readout for input instances of expression data, including uncertainty estimates. BC-Predict is freely available for non-commercial purposes at: https://apalania.shinyapps.io/BC-Predict.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"5 ","pages":"1644695"},"PeriodicalIF":3.9000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12488574/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2025.1644695","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
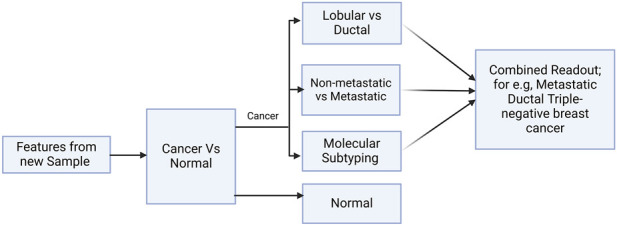
Introduction: Disease heterogeneity is the hallmark of breast cancer, which is the most common female malignancy. With a disturbing increase in mortality and disease burden, there remains a need for effective early-stage theragnostic and prognostic biomarkers. In this work, we improved on BrcaDx (https://apalania.shinyapps.io/brcadx/) for cancer vs control screening and examined a cluster of adjoining learning problems in breast cancer heterogeneity: (i) identification of metastatic cancers; (ii) molecular subtyping (TNBC, HER2, or luminal); and (iii) histological subtyping (invasive ductal or invasive lobular).
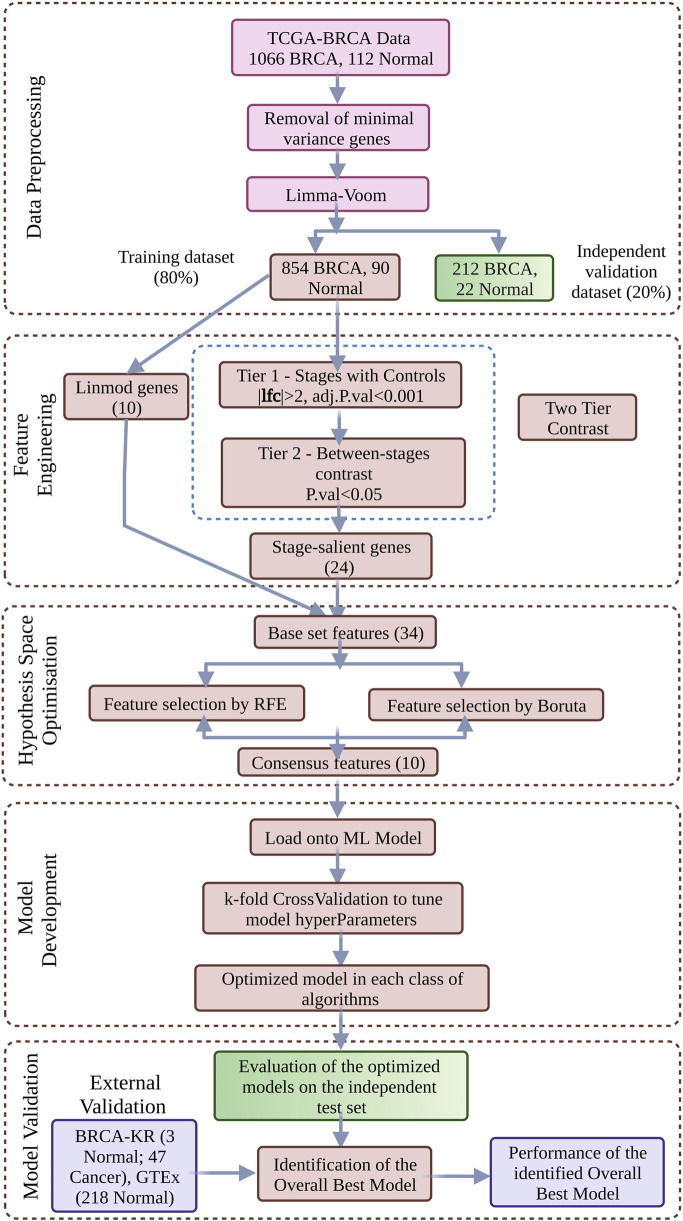
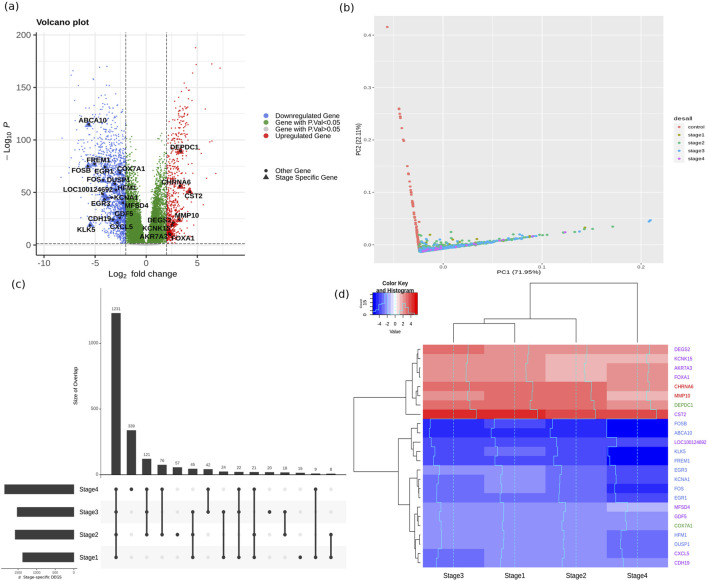
Methods: We analyzed the transcriptomic profiles of breast cancer patients from public-domain databases such as the TCGA using stage-encoded problem-specific statistical models of gene expression and unveiled stage-salient and progression-significant genes. Using a consensus approach, we identified potential machine learning features, and considered six model classes for each learning problem, with hyperparameter optimization on a training dataset and evaluation on a holdout test dataset. A nested approach enabled us to identify the best model class for each learning problem.
Results: External validation of the best models yielded balanced accuracies of 97.42% for cancer vs normal; 88.22% for metastatic v/s non metastatic; 88.79% for ternary molecular subtyping; and ensemble accuracy of 94.23% for histological subtyping. The model for molecular subtyping was validated on a 26-sample TNBC-only out-of-distribution cohort, yielding 25 correct predictions. We performed a late integration of multi-omics datasets by validating the feature space used in each problem with miRNA profiles, methylation profiles, and commercial breast cancer panels.
Discussion: Pending prospective studies, we have translated the models into BC-Predict that forks the best models developed for each problem in a unified interface and provides a complete readout for input instances of expression data, including uncertainty estimates. BC-Predict is freely available for non-commercial purposes at: https://apalania.shinyapps.io/BC-Predict.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


