{"title":"The Role of DFT Calculations in μ+SR Studies of Battery Materials","authors":"Hiroto Ohta*, and , Jun Sugiyama*, ","doi":"10.1021/acs.jpcc.5c03880","DOIUrl":null,"url":null,"abstract":"<p >Internal nuclear magnetic fields in various battery materials have been predicted using density functional theory (DFT) calculations to interpret the μ<sup>+</sup>SR results, particularly for identifying the diffusing species responsible for the dynamic behavior observed. In materials where Li<sup>+</sup> and Na<sup>+</sup> ions are mobile, these cations readily change positions to minimize electrostatic repulsion with the implanted μ<sup>+</sup>. As a result, the μ<sup>+</sup> sits at the bottom of a deep potential well, stabilizing itself through a “self-trapping″ effect, making it a stable observer for detecting ion diffusion in battery materials. In contrast, in many metals and oxides, the implanted μ<sup>+</sup> diffuses even at low temperatures. In these materials, the local lattice distortion caused by the implanted μ<sup>+</sup> is relatively small compared to that in battery materials. To assess the stability of the implanted μ<sup>+</sup>, we propose a ratio between the measured nuclear magnetic field distribution width (Δ<sup>exp</sup>) and the DFT-predicted value without lattice relaxation (Δ<sup>min</sup>), namely, Δ<sup>exp</sup>/Δ<sup>min</sup>, as an indicator of whether cations or μ<sup>+</sup> are diffusing. This indicator provides a comprehensive understanding of the diffusive behavior detected with μ<sup>+</sup>SR in various materials, including battery materials, metals, and other oxides.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 41","pages":"18406–18416"},"PeriodicalIF":3.2000,"publicationDate":"2025-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03880","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
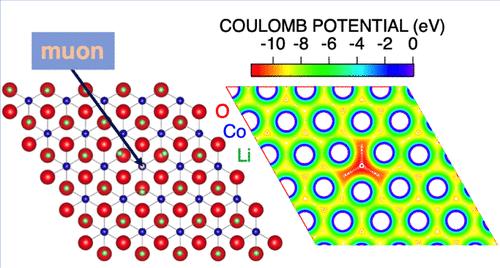
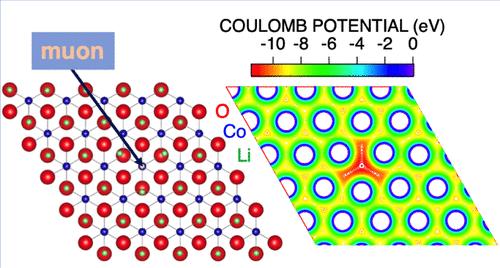
Internal nuclear magnetic fields in various battery materials have been predicted using density functional theory (DFT) calculations to interpret the μ+SR results, particularly for identifying the diffusing species responsible for the dynamic behavior observed. In materials where Li+ and Na+ ions are mobile, these cations readily change positions to minimize electrostatic repulsion with the implanted μ+. As a result, the μ+ sits at the bottom of a deep potential well, stabilizing itself through a “self-trapping″ effect, making it a stable observer for detecting ion diffusion in battery materials. In contrast, in many metals and oxides, the implanted μ+ diffuses even at low temperatures. In these materials, the local lattice distortion caused by the implanted μ+ is relatively small compared to that in battery materials. To assess the stability of the implanted μ+, we propose a ratio between the measured nuclear magnetic field distribution width (Δexp) and the DFT-predicted value without lattice relaxation (Δmin), namely, Δexp/Δmin, as an indicator of whether cations or μ+ are diffusing. This indicator provides a comprehensive understanding of the diffusive behavior detected with μ+SR in various materials, including battery materials, metals, and other oxides.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


