Electronic, structure, mechanical, optical and thermodynamic properties of double perovskite oxides X2ZrTeO6 (X =Cs, Rb, K): A first-principles study
IF 3.1
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Double perovskite compounds remain a central focus in materials research. We investigate the physical properties of X2ZrTeO6 (X = Cs, Rb, K) using first-principles calculations. Our results demonstrate the absence of imaginary frequencies in their phonon spectra, which satisfies the Born-Huang stability criteria and confirms their robust thermodynamic and kinetic stability. Mechanical stability is further verified through elastic constant analysis, with all compounds meeting the criteria: C11 > 0, C44 > 0, C11-C12 > 0, and C11 + 2C12 > 0. Electronic structure calculations reveal indirect bandgap semiconducting behavior, with HSE06-corrected band gaps of 3.002 eV (Cs), 3.550 eV (Rb), and 3.877 eV (K). Density of states analysis identifies O-p and Zr-d orbitals dominating the valence band maximum and O-p and Te-s states constituting the conduction band minimum, highlighting their role in charge transport. To assess the potential for optoelectronic applications, we calculate key optical properties, including dielectric functions, optical conductivity exceeding 8000 S cm−1 in the UV range. Thermodynamic studies indicate that the heat capacity approaches the Dulong-Petit limit near 800 K, consistent with high-temperature stability. The combination of stability, tunable bandgap, and strong UV absorption (peak at 5–15 eV) suggests promise for ultraviolet optoelectronic applications.
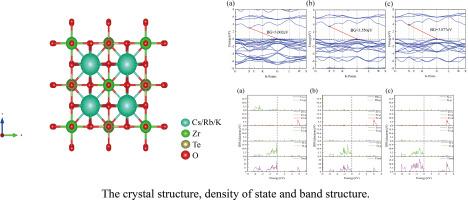
双钙钛矿氧化物X2ZrTeO6 (X =Cs, Rb, K)的电子、结构、机械、光学和热力学性质的第一性原理研究
双钙钛矿化合物一直是材料研究的热点。我们用第一性原理计算研究了X2ZrTeO6 (X = Cs, Rb, K)的物理性质。我们的研究结果表明,它们的声子谱中没有虚频率,这满足Born-Huang稳定性准则,并证实了它们的强大的热力学和动力学稳定性。通过弹性常数分析进一步验证了机械稳定性,所有化合物均满足标准:C11 >; 0, C44 > 0, C11- c12 > 0, C11 + 2C12 > 0。电子结构计算显示间接带隙半导体行为,hse06校正带隙为3.002 eV (Cs), 3.550 eV (Rb)和3.877 eV (K)。态密度分析发现O-p和Zr-d轨道主导价带最大值,O-p和Te-s轨道构成导带最小,突出了它们在电荷输运中的作用。为了评估光电子应用的潜力,我们计算了关键的光学性质,包括介电函数,在UV范围内超过8000 S cm−1的光电导率。热力学研究表明,热容在800k附近接近Dulong-Petit极限,符合高温稳定性。稳定性、可调带隙和强紫外吸收(峰值在5-15 eV)的结合表明了紫外光电应用的前景。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemical Physics Letters
化学-物理:原子、分子和化学物理
CiteScore
5.70
自引率
3.60%
发文量
798
审稿时长
33 days
期刊介绍:
Chemical Physics Letters has an open access mirror journal, Chemical Physics Letters: X, sharing the same aims and scope, editorial team, submission system and rigorous peer review.
Chemical Physics Letters publishes brief reports on molecules, interfaces, condensed phases, nanomaterials and nanostructures, polymers, biomolecular systems, and energy conversion and storage.
Criteria for publication are quality, urgency and impact. Further, experimental results reported in the journal have direct relevance for theory, and theoretical developments or non-routine computations relate directly to experiment. Manuscripts must satisfy these criteria and should not be minor extensions of previous work.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


