Optimizing the basis set extrapolation parameter for weak interaction energy calculations using density functional theory
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Accurate computation of weak interaction energies remains challenging due to basis set superposition error (BSSE) and the need for diffuse functions. While the counterpoise (CP) method is commonly used to correct BSSE, it increases computational cost and complexity. This work assesses the exponential-square-root extrapolation scheme for density functional theory (DFT) calculations of weak interactions. An optimized extrapolation exponent (α = 5.674) was obtained from a training set of 57 complexes and applied to a two-point extrapolation using def2-SVP and def2-TZVPP basis sets. The resulting interaction energies closely match those from CP-corrected ma-TZVPP calculations, with a mean relative error of ~2%. Although slightly less accurate than CP-corrected values, the extrapolation method requires only about half the computational time. Our results also show that diffuse functions are unnecessary for neutral systems when using CP with def2-TZVPP. Validation on the S66, L7, and NIAR20 benchmark sets and across other functionals confirms the method's robustness and transferability.
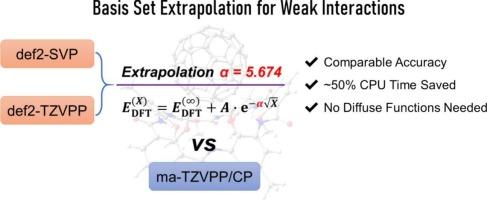
利用密度泛函理论优化弱相互作用能计算的基集外推参数
由于基集叠加误差(BSSE)和对扩散函数的需要,弱相互作用能的精确计算仍然是一个挑战。通常采用平衡法校正BSSE,但这种方法增加了计算成本和计算复杂度。本研究评估了密度泛函理论(DFT)计算弱相互作用的指数平方根外推方案。从57个复合物的训练集中得到优化的外推指数(α = 5.674),并将其应用于def2-SVP和def2-TZVPP基集的两点外推。得到的相互作用能与cp校正后的ma-TZVPP计算结果非常接近,平均相对误差约为2%。虽然外推法的精度略低于cp校正值,但它只需要大约一半的计算时间。我们的结果还表明,当使用具有def2-TZVPP的CP时,中性系统不需要扩散函数。在S66、L7和NIAR20基准集和其他功能上的验证证实了该方法的鲁棒性和可移植性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


