{"title":"vcfsim: flexible simulation of all-sites VCFs with missing data.","authors":"Paimon Goulart, Kieran Samuk","doi":"10.1101/2025.01.29.635540","DOIUrl":null,"url":null,"abstract":"<p><strong>Background |: </strong>VCFs are the most widely used data format for encoding genetic variation. By design, standard VCFs do not include data from sites where all individuals are homozygous for the reference allele (\"invariant sites\") and thus do not differentiate these from sites where data are completely missing. However, missing data are a key feature of biological datasets across all domains of genomics, and many recent studies have shown that missing data can introduce a variety of statistical biases in the estimation of key population genetic parameters. A solution to this limitation is to include invariant sites in a standard VCF, creating an \"all-sites VCF\", exposing missing and invariant sites explicitly. One hurdle to the wider adoption of all-sites VCFs is a reliable parameterized simulation framework for generating biologically realistic all-sites VCFs.</p><p><strong>Results |: </strong>Here, we introduce an open-source command line tool, <i>vcfsim</i>, that interfaces with the popular coalescent simulation platform <i>msprime</i> and provides convenience functions for simulating all-sites VCFs with variable levels of ploidy and missing data. We show that the post-processed VCFs generated using <i>vcfsim</i> align precisely with population genetic expectations (i.e. are statistically identical to raw <i>msprime</i> output), accurately introduce missing data, and permit the simulation of data with varying ploidy levels, including the simulation of intraindividual ploidy variation (e.g. heterogametic sex chromosomes) and population structures.</p><p><strong>Conclusions |: </strong>Our results <i>vcfsim</i> is a useful and easy-to-use tool for the benchmarking of new software tools, performing population genetic inference, training of machine learning models, and the exploration of the effects of missing data in genomics data sets.</p>","PeriodicalId":519960,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12486129/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2025.01.29.635540","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
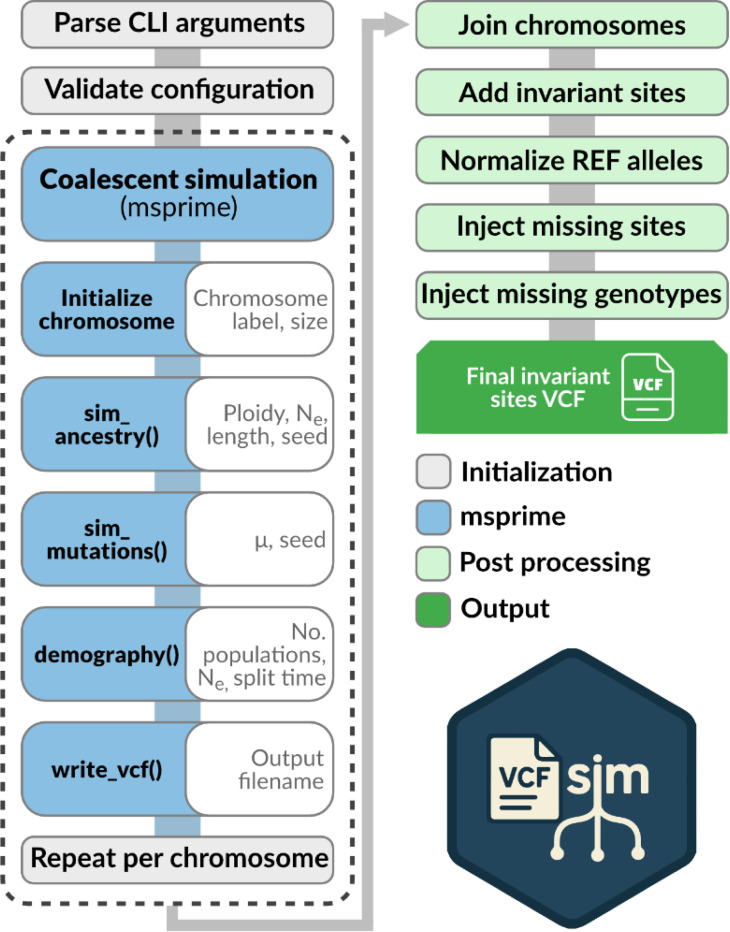
Background |: VCFs are the most widely used data format for encoding genetic variation. By design, standard VCFs do not include data from sites where all individuals are homozygous for the reference allele ("invariant sites") and thus do not differentiate these from sites where data are completely missing. However, missing data are a key feature of biological datasets across all domains of genomics, and many recent studies have shown that missing data can introduce a variety of statistical biases in the estimation of key population genetic parameters. A solution to this limitation is to include invariant sites in a standard VCF, creating an "all-sites VCF", exposing missing and invariant sites explicitly. One hurdle to the wider adoption of all-sites VCFs is a reliable parameterized simulation framework for generating biologically realistic all-sites VCFs.
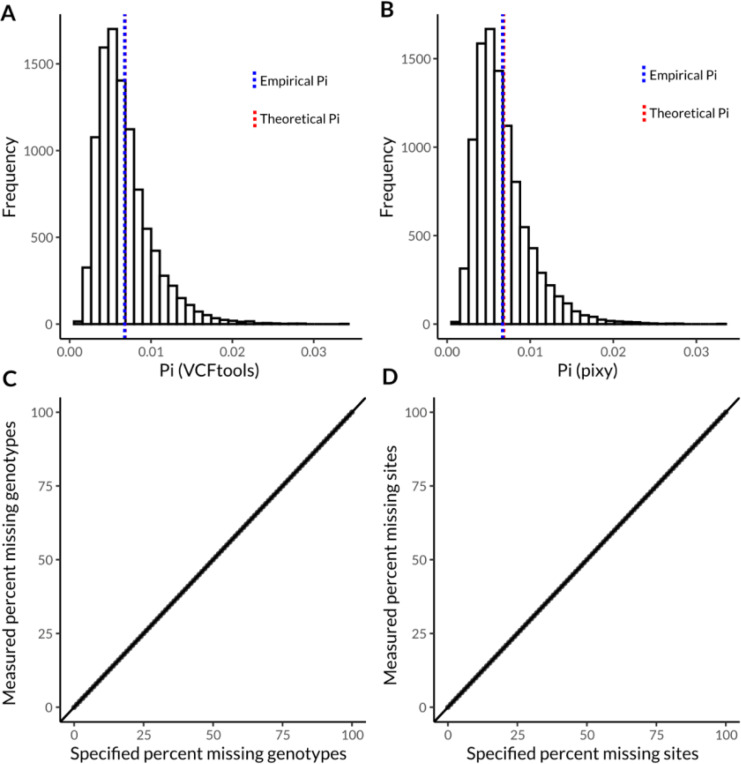
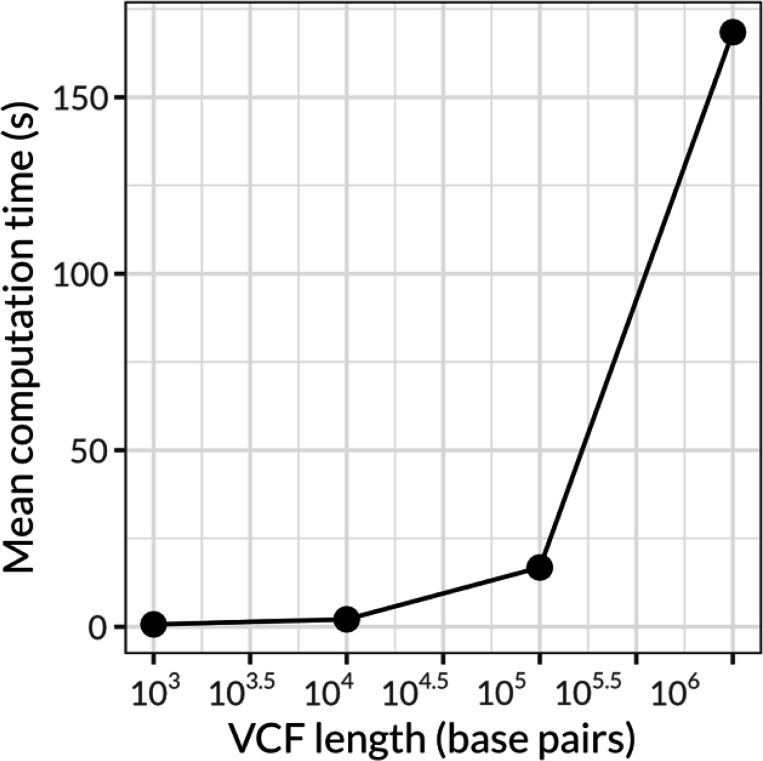
Results |: Here, we introduce an open-source command line tool, vcfsim, that interfaces with the popular coalescent simulation platform msprime and provides convenience functions for simulating all-sites VCFs with variable levels of ploidy and missing data. We show that the post-processed VCFs generated using vcfsim align precisely with population genetic expectations (i.e. are statistically identical to raw msprime output), accurately introduce missing data, and permit the simulation of data with varying ploidy levels, including the simulation of intraindividual ploidy variation (e.g. heterogametic sex chromosomes) and population structures.
Conclusions |: Our results vcfsim is a useful and easy-to-use tool for the benchmarking of new software tools, performing population genetic inference, training of machine learning models, and the exploration of the effects of missing data in genomics data sets.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


