Hongxia Fu, Lu Han, Xianhong Liu, Bin He, Pei He, Junjian Hu
{"title":"A Novel Missense Variant of the ABCD1 Gene in X-Linked Adrenoleukodystrophy in Chinese Family.","authors":"Hongxia Fu, Lu Han, Xianhong Liu, Bin He, Pei He, Junjian Hu","doi":"10.1002/mgg3.70148","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>We identified a novel ABCD1 variant (c.773T>G, p.Leu258Arg, NM_000033.4) in a Chinese pedigree affected by X-linked adrenoleukodystrophy (X-ALD). This missense variant in exon 1 is predicted to be pathogenic and likely constitutes the genetic basis of the disease phenotype in this family.</p><p><strong>Methods: </strong>ABCD1 gene sequencing was performed in the Chinese pedigree. The pathogenicity of identified variants was assessed using computational prediction tools. Subcellular localization studies were conducted, and very-long-chain fatty acid (VLCFA) levels were quantified in patient-derived samples.</p><p><strong>Results: </strong>Sequencing analysis identified a hemizygous missense variant in the ABCD1 gene (c.773T>G; p.Leu258Arg). In silico pathogenicity prediction using SIFT and PolyPhen-2 algorithms classified the p.Leu258Arg substitution as deleterious. Functional characterization revealed that the p.Leu258Arg variant impairs the peroxisomal membrane localization of the ABCD1 protein. Consistent with the established role of ABCD1 in peroxisomal β-oxidation, individuals harboring this variant exhibited significantly elevated serum levels of VLCFA. Specifically, the C26:0/C22:0 ratio was elevated 2.8-fold compared to control values, confirming impaired VLCFA metabolism.</p><p><strong>Conclusion: </strong>In accordance with the \"Standards and Guidelines for the Interpretation of Sequence Variants\" established by the American College of Medical Genetics and Genomics (ACMG), we assessed the pathogenicity of the novel ABCD1 gene variant c.773T>G. This variant meets the following ACMG evidence criteria: PM1 (located within a critical functional domain or mutational hotspot known to lack benign variation); PM2 (absent or observed at very low frequency in population databases e.g., gnomAD, EXAC, 1000 Genomes); PP3 (multiple in silico prediction tools consistently suggest a deleterious effect on the gene or gene product). Integrating this evidence (PM1 + PM2 + PP3), the variant is classified as likely pathogenic based on ACMG guidelines. Experimental data from this study further substantiate the pathogenicity of the c.773T>G variant located in exon 1 of the ABCD1 gene. This finding broadens the spectrum of known pathogenic mutations in ABCD1 associated with X-ALD and provides crucial information for the molecular diagnosis of affected patients.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 10","pages":"e70148"},"PeriodicalIF":1.6000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12492069/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70148","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
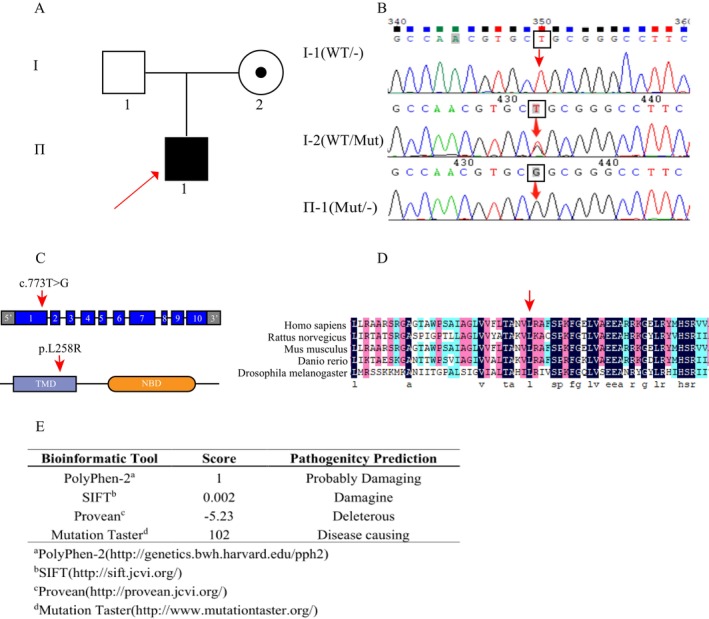
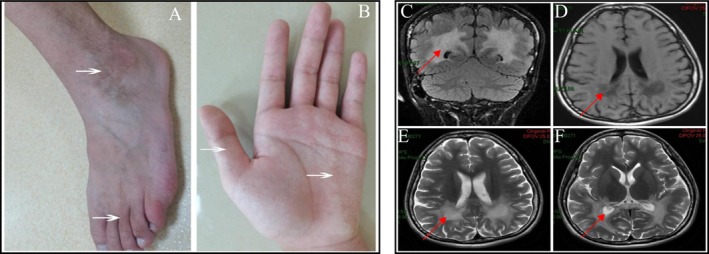
Background: We identified a novel ABCD1 variant (c.773T>G, p.Leu258Arg, NM_000033.4) in a Chinese pedigree affected by X-linked adrenoleukodystrophy (X-ALD). This missense variant in exon 1 is predicted to be pathogenic and likely constitutes the genetic basis of the disease phenotype in this family.
Methods: ABCD1 gene sequencing was performed in the Chinese pedigree. The pathogenicity of identified variants was assessed using computational prediction tools. Subcellular localization studies were conducted, and very-long-chain fatty acid (VLCFA) levels were quantified in patient-derived samples.
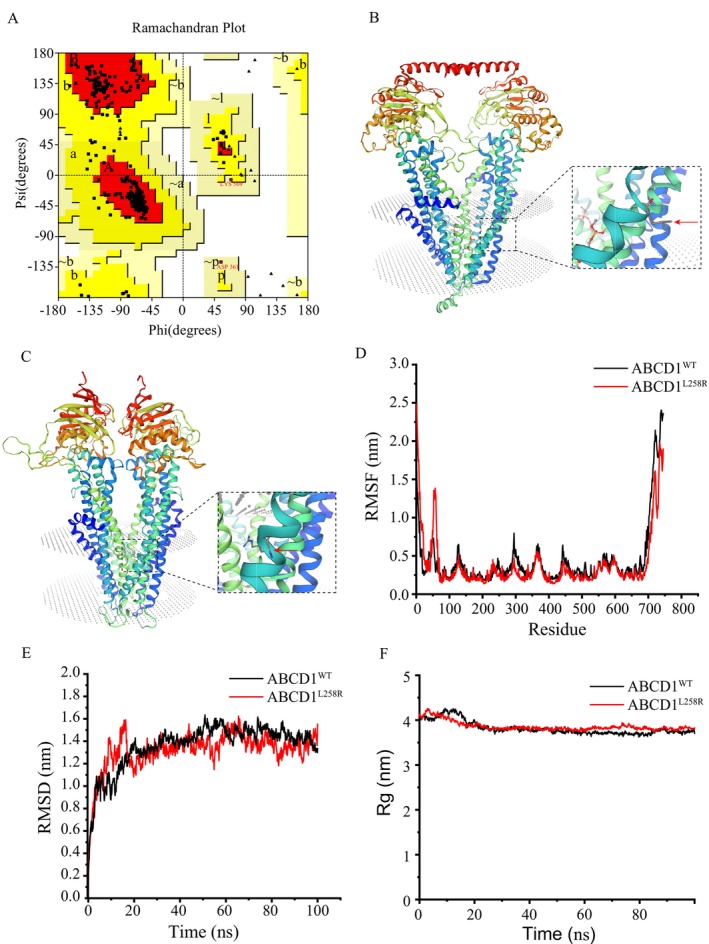
Results: Sequencing analysis identified a hemizygous missense variant in the ABCD1 gene (c.773T>G; p.Leu258Arg). In silico pathogenicity prediction using SIFT and PolyPhen-2 algorithms classified the p.Leu258Arg substitution as deleterious. Functional characterization revealed that the p.Leu258Arg variant impairs the peroxisomal membrane localization of the ABCD1 protein. Consistent with the established role of ABCD1 in peroxisomal β-oxidation, individuals harboring this variant exhibited significantly elevated serum levels of VLCFA. Specifically, the C26:0/C22:0 ratio was elevated 2.8-fold compared to control values, confirming impaired VLCFA metabolism.
Conclusion: In accordance with the "Standards and Guidelines for the Interpretation of Sequence Variants" established by the American College of Medical Genetics and Genomics (ACMG), we assessed the pathogenicity of the novel ABCD1 gene variant c.773T>G. This variant meets the following ACMG evidence criteria: PM1 (located within a critical functional domain or mutational hotspot known to lack benign variation); PM2 (absent or observed at very low frequency in population databases e.g., gnomAD, EXAC, 1000 Genomes); PP3 (multiple in silico prediction tools consistently suggest a deleterious effect on the gene or gene product). Integrating this evidence (PM1 + PM2 + PP3), the variant is classified as likely pathogenic based on ACMG guidelines. Experimental data from this study further substantiate the pathogenicity of the c.773T>G variant located in exon 1 of the ABCD1 gene. This finding broadens the spectrum of known pathogenic mutations in ABCD1 associated with X-ALD and provides crucial information for the molecular diagnosis of affected patients.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


