{"title":"In silico identification of anticancer flavonoids as dengue virus replication inhibitors: a molecular docking and simulation approach","authors":"Soumendu Patra, Arindam Paul, Harshita Shand, Sayan Ghosal, Suvankar Ghorai","doi":"10.1007/s00894-025-06516-3","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Dengue, a mosquito-borne viral disease endemic to over 100 countries, poses a serious health risk to people living in tropical regions. The viral non-structural proteins NS3 (helicase) and NS5 (RNA-dependent RNA polymerase) are critical targets for antiviral drug development. Several natural and synthetic compounds have been tried against this for the screening of antiviral inhibitor(s) but so far limited success has been achieved. In this study, we have investigated how natural products interact with and destabilize NS3 and NS5. We used in silico methods to screen new potential NS3 and NS5 inhibitors from various anti-cancer flavonoid compounds that previously showed anti-cancer properties in vitro. A virtual screening was conducted on 329 anti-cancer flavonoid compounds, selecting 190 compounds based on Lipinski’s rule of five, the Muegge filter, the Ghose filter, and the Veber filter. Molecular docking techniques allowed us to identify Artobiloxanthone (PubChem CID: 46887866) as the most effective binder for NS3, while Glabridin (PubChem CID: 124052) emerged as the most effective binder for NS5. These leading candidates demonstrated favorable ADMET profiles. Additionally, molecular dynamics (MD) simulations of up to 1000-ns showcased stable protein–ligand interactions, with convergence achieved at 200 ns for NS3 and 470 ns for NS5. The structural stability of the complexes was validated by analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding (H-bonding). Binding affinity between the ligand and target proteins was further validated by binding free energy calculations using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach, providing a robust estimation of interaction stability. These findings highlight the potential of Artobiloxanthone and Glabridin as promising inhibitors of dengue virus replication.</p><h3>Methods</h3><p>The Chimera v1.11.2 program was employed for protein optimization, while PyRx 0.8 served for molecular docking. Protein structures were converted into.pdbqt format, and ligands were energy-minimized using the MMFF94 force field and conjugate gradient optimization. Visualization was conducted using BIOVIA Discovery Studio Visualizer and PyMOL. The ADMET properties of the top hits were predicted using the pkCSM and ProTox-II platforms. To address missing residues in the crystal structures, AlphaFold2 was used to predict full-length protein models. MD simulations were performed with the GROMACS 2024.5 package, utilizing the AMBER-f99SB-ILDN force field, the TIP3P water model, and a 120 mM NaCl concentration. Equilibration was achieved through V-rescale and C-rescale thermostats, followed by Parrinello–Rahman barostat production runs. Analysis of the MD trajectories included RMSD, RMSF, Rg, SASA, H-bonding, and MM-PBSA utilizing GROMACS tools, gmx_MMPBSA, VMD, and MDAnalysis.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06516-3","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
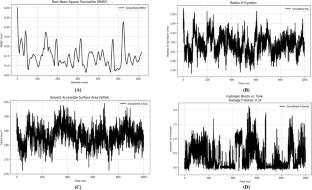
Dengue, a mosquito-borne viral disease endemic to over 100 countries, poses a serious health risk to people living in tropical regions. The viral non-structural proteins NS3 (helicase) and NS5 (RNA-dependent RNA polymerase) are critical targets for antiviral drug development. Several natural and synthetic compounds have been tried against this for the screening of antiviral inhibitor(s) but so far limited success has been achieved. In this study, we have investigated how natural products interact with and destabilize NS3 and NS5. We used in silico methods to screen new potential NS3 and NS5 inhibitors from various anti-cancer flavonoid compounds that previously showed anti-cancer properties in vitro. A virtual screening was conducted on 329 anti-cancer flavonoid compounds, selecting 190 compounds based on Lipinski’s rule of five, the Muegge filter, the Ghose filter, and the Veber filter. Molecular docking techniques allowed us to identify Artobiloxanthone (PubChem CID: 46887866) as the most effective binder for NS3, while Glabridin (PubChem CID: 124052) emerged as the most effective binder for NS5. These leading candidates demonstrated favorable ADMET profiles. Additionally, molecular dynamics (MD) simulations of up to 1000-ns showcased stable protein–ligand interactions, with convergence achieved at 200 ns for NS3 and 470 ns for NS5. The structural stability of the complexes was validated by analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding (H-bonding). Binding affinity between the ligand and target proteins was further validated by binding free energy calculations using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach, providing a robust estimation of interaction stability. These findings highlight the potential of Artobiloxanthone and Glabridin as promising inhibitors of dengue virus replication.
Methods
The Chimera v1.11.2 program was employed for protein optimization, while PyRx 0.8 served for molecular docking. Protein structures were converted into.pdbqt format, and ligands were energy-minimized using the MMFF94 force field and conjugate gradient optimization. Visualization was conducted using BIOVIA Discovery Studio Visualizer and PyMOL. The ADMET properties of the top hits were predicted using the pkCSM and ProTox-II platforms. To address missing residues in the crystal structures, AlphaFold2 was used to predict full-length protein models. MD simulations were performed with the GROMACS 2024.5 package, utilizing the AMBER-f99SB-ILDN force field, the TIP3P water model, and a 120 mM NaCl concentration. Equilibration was achieved through V-rescale and C-rescale thermostats, followed by Parrinello–Rahman barostat production runs. Analysis of the MD trajectories included RMSD, RMSF, Rg, SASA, H-bonding, and MM-PBSA utilizing GROMACS tools, gmx_MMPBSA, VMD, and MDAnalysis.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


