Borja Aldeguer-Riquelme, Luis M Rodriguez-R, Konstantinos T Konstantinidis
{"title":"Differences in metagenome coverage may confound abundance-based and diversity conclusions and how to deal with them.","authors":"Borja Aldeguer-Riquelme, Luis M Rodriguez-R, Konstantinos T Konstantinidis","doi":"10.1093/ismeco/ycaf140","DOIUrl":null,"url":null,"abstract":"<p><p>The importance of rarefying ecological or amplicon sequencing data to a standardized level of diversity coverage for reliable diversity comparisons across samples is well recognized. However, the importance of diversity coverage, i.e. the fraction of the genomic diversity of a sample sequenced, in comparative shotgun metagenomic studies remains frequently overlooked. Using both <i>in silico</i> and natural metagenomes from a wide range of environments, we demonstrate that uneven metagenome coverage can result in misleading biological conclusions, particularly for identifying differentially abundant features, i.e. groups of genes or genomes assigned to the same protein family or taxonomic rank, respectively, and for comparing diversity between samples. The main underlying cause is that not all members of a feature may be detectable, and thus counted, across such unevenly covered metagenomes depending on the sequencing effort applied and the underlying member-abundance curves. Unfortunately, 99.5% of previous comparative metagenomic studies have overlooked this metric, suggesting that their reported results might be misleading. We show that achieving high Nonpareil coverage (≥0.9), a metric that estimates metagenome diversity coverage, is the most reliable strategy to mitigate this issue. When high Nonpareil coverage is not achievable, such as for highly diverse and complex samples like soils, we show that standardizing (or subsampling) metagenomic datasets to the same Nonpareil coverage, rather than sequencing effort, prior to comparative analysis provides for more accurate results. We provide a set of practical recommendations and the corresponding Python scripts to help researchers to assess and standardize metagenome diversity coverage for their comparative analyses.</p>","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":"5 1","pages":"ycaf140"},"PeriodicalIF":6.1000,"publicationDate":"2025-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12477595/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/ismeco/ycaf140","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
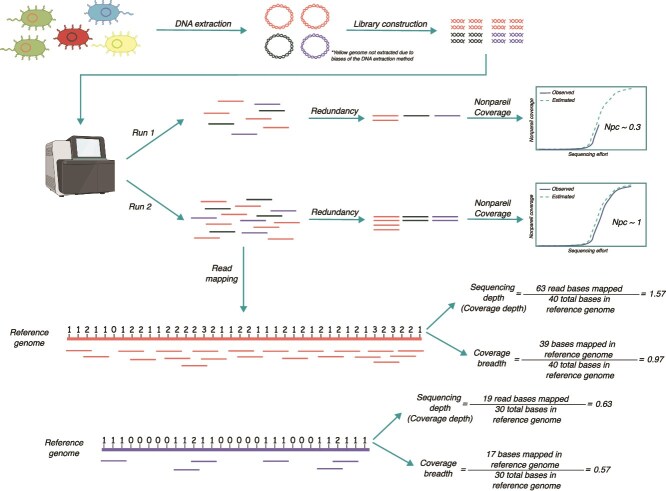
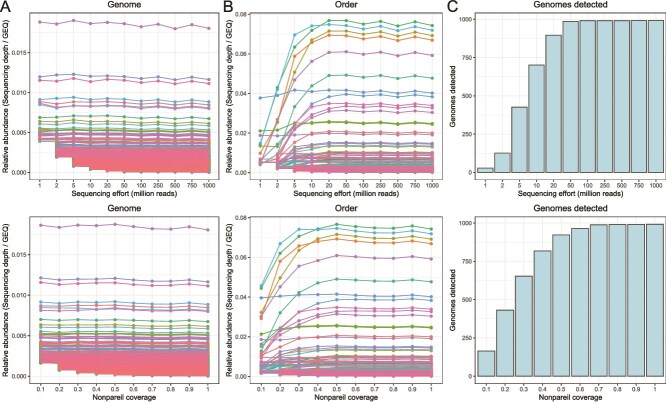
The importance of rarefying ecological or amplicon sequencing data to a standardized level of diversity coverage for reliable diversity comparisons across samples is well recognized. However, the importance of diversity coverage, i.e. the fraction of the genomic diversity of a sample sequenced, in comparative shotgun metagenomic studies remains frequently overlooked. Using both in silico and natural metagenomes from a wide range of environments, we demonstrate that uneven metagenome coverage can result in misleading biological conclusions, particularly for identifying differentially abundant features, i.e. groups of genes or genomes assigned to the same protein family or taxonomic rank, respectively, and for comparing diversity between samples. The main underlying cause is that not all members of a feature may be detectable, and thus counted, across such unevenly covered metagenomes depending on the sequencing effort applied and the underlying member-abundance curves. Unfortunately, 99.5% of previous comparative metagenomic studies have overlooked this metric, suggesting that their reported results might be misleading. We show that achieving high Nonpareil coverage (≥0.9), a metric that estimates metagenome diversity coverage, is the most reliable strategy to mitigate this issue. When high Nonpareil coverage is not achievable, such as for highly diverse and complex samples like soils, we show that standardizing (or subsampling) metagenomic datasets to the same Nonpareil coverage, rather than sequencing effort, prior to comparative analysis provides for more accurate results. We provide a set of practical recommendations and the corresponding Python scripts to help researchers to assess and standardize metagenome diversity coverage for their comparative analyses.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


