Phthalimide–benzoic acid hybrids as potent aldose reductase inhibitors: Synthesis, enzymatic kinetics, and in silico characterization
IF 3
3区 医学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Aldose reductase (ALR2; AKR1B1) is implicated in hyperglycemia-driven tissue injury and remains a tractable enzymatic target. We developed a concise, chromatography-free two-step route to phthalimide–benzoic acid hybrids (5a–5m) and profiled their biochemical activity against human ALR2. Across the series, halogenated analogs were most active, with the para-bromophenyl derivative 5d emerging as the top hit (KI = 7.56 nM). Steady-state kinetic analysis indicated a competitive inhibition mechanism. Molecular docking to the ALR2 active site (PDB 4JIR), supported by MM-GBSA rescoring, yielded a catalytically consistent binding mode featuring hydrogen-bonding within the anion-binding region (Tyr48, His110) and complementary hydrophobic contacts (Trp111, Trp219), with Cys298 contributing as a proximal hydrophobic contact. In cell-based assays (A549, Hep3B, L929), the compounds generally showed low intrinsic cytotoxicity at the tested concentrations, suggesting a favorable preliminary safety margin aligned with their ALR2-directed pharmacology. In silico ADME/Tox assessments further supported oral drug-likeness. Overall, these results identify phthalimide–benzoic acid hybrids as tractable ALR2 inhibitor scaffolds that combine potent biochemical inhibition with a competitive kinetic profile and encouraging early safety signals, warranting in vivo evaluation and SAR-guided optimization.
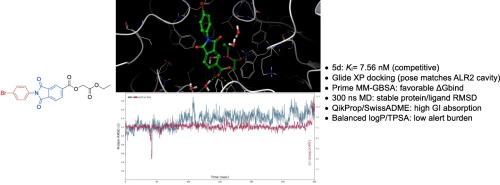
邻苯二胺-苯甲酸杂合体作为强效醛糖还原酶抑制剂:合成、酶动力学和硅表征。
醛糖还原酶(ALR2; AKR1B1)与高血糖驱动的组织损伤有关,并且仍然是一个易于处理的酶靶点。我们开发了一种简洁、免色谱的两步法合成邻苯二胺-苯甲酸杂合体(5a-5m),并分析了它们对人类ALR2的生化活性。在整个系列中,卤化类似物的活性最高,对溴苯基衍生物5d的活性最高(KI = 7.56 nM)。稳态动力学分析表明存在竞争性抑制机制。在MM-GBSA的支持下,与ALR2活性位点(PDB 4JIR)的分子对接产生了一种催化一致的结合模式,即阴离子结合区域(Tyr48, His110)内的氢键和互补的疏水接触(Trp111, Trp219),其中Cys298作为近端疏水接触。在基于细胞的实验(A549, Hep3B, L929)中,这些化合物在测试浓度下通常表现出较低的内在细胞毒性,这表明它们具有良好的初步安全边际,与它们的alr2导向药理学一致。计算机ADME/Tox评估进一步支持口服药物相似性。总的来说,这些结果确定了邻苯二胺-苯甲酸杂交种是易于处理的ALR2抑制剂支架,它结合了强大的生化抑制和竞争性动力学特征,并鼓励早期安全信号,保证了体内评估和sar引导的优化。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Bioorganic & Medicinal Chemistry
医学-生化与分子生物学
CiteScore
6.80
自引率
2.90%
发文量
413
审稿时长
17 days
期刊介绍:
Bioorganic & Medicinal Chemistry provides an international forum for the publication of full original research papers and critical reviews on molecular interactions in key biological targets such as receptors, channels, enzymes, nucleotides, lipids and saccharides.
The aim of the journal is to promote a better understanding at the molecular level of life processes, and living organisms, as well as the interaction of these with chemical agents. A special feature will be that colour illustrations will be reproduced at no charge to the author, provided that the Editor agrees that colour is essential to the information content of the illustration in question.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


