Jang Mok Yoo, , , Mathew Chow, , , Eno Paenurk, , and , Sharon Hammes-Schiffer*,
{"title":"Reliable pKa Prediction through Efficient Incorporation of Anharmonicity within the Nuclear–Electronic Orbital Framework","authors":"Jang Mok Yoo, , , Mathew Chow, , , Eno Paenurk, , and , Sharon Hammes-Schiffer*, ","doi":"10.1021/jacs.5c11332","DOIUrl":null,"url":null,"abstract":"<p >Accurate p<i>K</i><sub>a</sub> prediction is critical for understanding chemical reactivity and molecular properties across a wide range of applications. Computational approaches usually invoke a harmonic treatment of the vibrational modes for zero-point energies, as well as thermal and entropic contributions. Herein, we present a general protocol for relative p<i>K</i><sub>a</sub> prediction that incorporates the significant anharmonic effects using nuclear–electronic orbital (NEO) theory. This protocol is validated against experimental data for a range of molecules in acetonitrile, including protonated nitrogen bases, nitrophenols, anilines, and diamines, as well as cobalt electrocatalysts. For simple acids, the NEO approach offers only a slight improvement over conventional density functional theory with the standard harmonic vibrational treatment, whereas for hydrogen-bonded acids, the NEO approach offers more significantly improved performance at a comparable computational cost. This accessible methodology provides a practical route for accurate p<i>K</i><sub>a</sub> prediction in challenging systems and is extendable to related thermodynamic properties such as hydricities and proton-coupled redox potentials.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"147 40","pages":"36059–36065"},"PeriodicalIF":15.6000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.5c11332","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
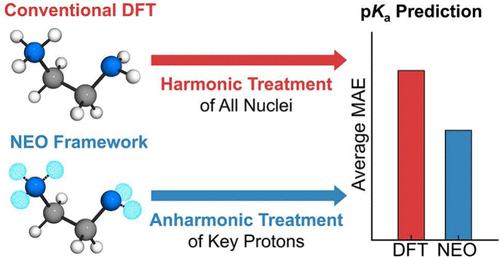
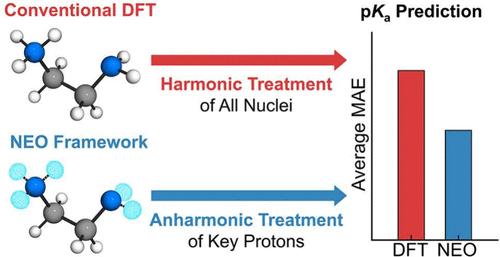
Accurate pKa prediction is critical for understanding chemical reactivity and molecular properties across a wide range of applications. Computational approaches usually invoke a harmonic treatment of the vibrational modes for zero-point energies, as well as thermal and entropic contributions. Herein, we present a general protocol for relative pKa prediction that incorporates the significant anharmonic effects using nuclear–electronic orbital (NEO) theory. This protocol is validated against experimental data for a range of molecules in acetonitrile, including protonated nitrogen bases, nitrophenols, anilines, and diamines, as well as cobalt electrocatalysts. For simple acids, the NEO approach offers only a slight improvement over conventional density functional theory with the standard harmonic vibrational treatment, whereas for hydrogen-bonded acids, the NEO approach offers more significantly improved performance at a comparable computational cost. This accessible methodology provides a practical route for accurate pKa prediction in challenging systems and is extendable to related thermodynamic properties such as hydricities and proton-coupled redox potentials.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


