Unveiling the Antidiabetic Potential of 6-Chloro-3-(2-substituted-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one Derivatives: Design, Synthesis, and Biological Evaluation Targeting Fructose 1,6-Bisphosphatase with In Silico ADMET and Docking Approaches
{"title":"Unveiling the Antidiabetic Potential of 6-Chloro-3-(2-substituted-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one Derivatives: Design, Synthesis, and Biological Evaluation Targeting Fructose 1,6-Bisphosphatase with In Silico ADMET and Docking Approaches","authors":"Sanjay D. Sawant, Vasundhara N. Sawant","doi":"10.1134/S1068162025600047","DOIUrl":null,"url":null,"abstract":"<p><b>Objective:</b> FBPase is considered a significant player in the middle phase of the gluconeogenesis pathway and an attractive target for improving hyperglycemia in T2DM patients. To date, several FBPase inhibitors for the management of diabetes have been reported; however, many of them are structurally similar to AMP, resulting in some off-target side effects. Therefore, efforts have been made to identify novel non-AMP-mimetic FBPase inhibitors without a phosphonate moiety. <b>Methods:</b> Compounds bearing a quinazoline scaffold linked to a thiazolidinone moiety were investigated as FBPase inhibitors in this study. The synthesis, molecular docking studies, pharmacokinetic predictions, toxicity profiles, and biological activities of quinazoline–thiazolidinone derivatives are reported. <i>In silico</i> approaches were thoroughly employed to understand the binding modes at the active site of FBPase. <b>Results and Discussion:</b> The results indicated that several compounds demonstrated significant antidiabetic activity, with the highest activity observed in compound (<b>S6</b>). An in-depth examination of structural variations and their biological activities revealed that (<b>S6</b>) is the most potent compound, with an IC<sub>50</sub> value of 0.004 ± 0.10 µM. <b>Conclusions:</b> Compound (<b>S6</b>) exhibited noteworthy hypoglycemic activity and can serve as a template for the future development of FBPase inhibitors. This work also demonstrates that orally bioavailable and effective FBPase inhibitors can be developed without employing prodrug strategies.</p>","PeriodicalId":758,"journal":{"name":"Russian Journal of Bioorganic Chemistry","volume":"51 5","pages":"2185 - 2204"},"PeriodicalIF":1.7000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of Bioorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S1068162025600047","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
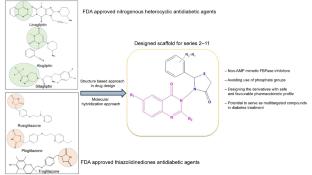
Objective: FBPase is considered a significant player in the middle phase of the gluconeogenesis pathway and an attractive target for improving hyperglycemia in T2DM patients. To date, several FBPase inhibitors for the management of diabetes have been reported; however, many of them are structurally similar to AMP, resulting in some off-target side effects. Therefore, efforts have been made to identify novel non-AMP-mimetic FBPase inhibitors without a phosphonate moiety. Methods: Compounds bearing a quinazoline scaffold linked to a thiazolidinone moiety were investigated as FBPase inhibitors in this study. The synthesis, molecular docking studies, pharmacokinetic predictions, toxicity profiles, and biological activities of quinazoline–thiazolidinone derivatives are reported. In silico approaches were thoroughly employed to understand the binding modes at the active site of FBPase. Results and Discussion: The results indicated that several compounds demonstrated significant antidiabetic activity, with the highest activity observed in compound (S6). An in-depth examination of structural variations and their biological activities revealed that (S6) is the most potent compound, with an IC50 value of 0.004 ± 0.10 µM. Conclusions: Compound (S6) exhibited noteworthy hypoglycemic activity and can serve as a template for the future development of FBPase inhibitors. This work also demonstrates that orally bioavailable and effective FBPase inhibitors can be developed without employing prodrug strategies.
期刊介绍:
Russian Journal of Bioorganic Chemistry publishes reviews and original experimental and theoretical studies on the structure, function, structure–activity relationships, and synthesis of biopolymers, such as proteins, nucleic acids, polysaccharides, mixed biopolymers, and their complexes, and low-molecular-weight biologically active compounds (peptides, sugars, lipids, antibiotics, etc.). The journal also covers selected aspects of neuro- and immunochemistry, biotechnology, and ecology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


