Stéphanie Chevalier, Julia Becker, Yujuan Gui, Vincent Noël, Cui Su, Sascha Jung, Laurence Calzone, Andrei Zinovyev, Antonio Del Sol, Jun Pang, Lasse Sinkkonen, Thomas Sauter, Loïc Paulevé
{"title":"Data-driven inference of Boolean networks from transcriptomes to predict cellular differentiation and reprogramming.","authors":"Stéphanie Chevalier, Julia Becker, Yujuan Gui, Vincent Noël, Cui Su, Sascha Jung, Laurence Calzone, Andrei Zinovyev, Antonio Del Sol, Jun Pang, Lasse Sinkkonen, Thomas Sauter, Loïc Paulevé","doi":"10.1038/s41540-025-00569-z","DOIUrl":null,"url":null,"abstract":"<p><p>Boolean networks provide robust, explainable, and predictive models of cellular dynamics, especially for cellular differentiation and fate decision processes. Yet, the construction of such models is extremely challenging, as it requires integrating prior knowledge with experimental observation of the transcriptome, potentially relating thousands of genes. We present a general methodology for integrating transcriptome data and prior knowledge on the underlying gene regulatory network in order to generate automatically ensembles of Boolean networks able to reproduce the modeled qualitative behavior. Our methodology builds on the software BoNesis, which implements the automatic construction of Boolean networks from a specification of their expected structural and dynamical properties. We show how to transform transcriptome data into such a qualitative specification, and then how to exploit the generated ensembles of Boolean networks for identifying families of candidate models, and for predicting robust cellular reprogramming targets. We illustrate the scalability and versatility of our overall approach with two applications: the modeling of hematopoiesis from single-cell RNA-Seq data, and modeling the differentiation of bone marrow stromal cells into adipocytes and osteoblasts from bulk RNA-seq time series data. For this latter case, we took advantage of ensemble modeling to predict combinations of reprogramming factors for trans-differentiation that are robust to model uncertainties due to variations in experimental replicates and choice of binarization method. Moreover, we performed an in silico assessment of the fidelity and efficiency of the reprogramming and conducted preliminary experimental validation.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"11 1","pages":"105"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12475257/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-025-00569-z","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
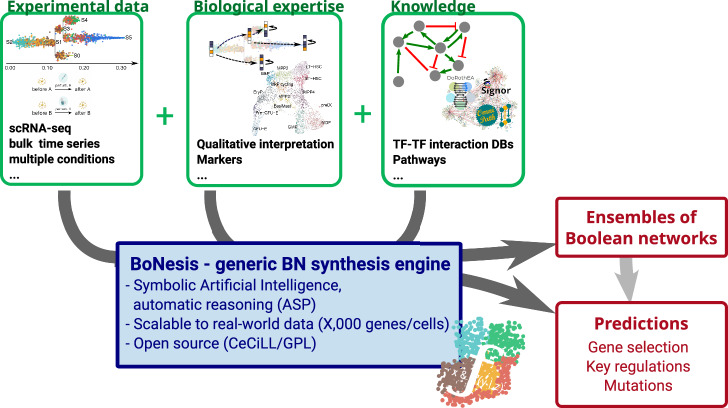
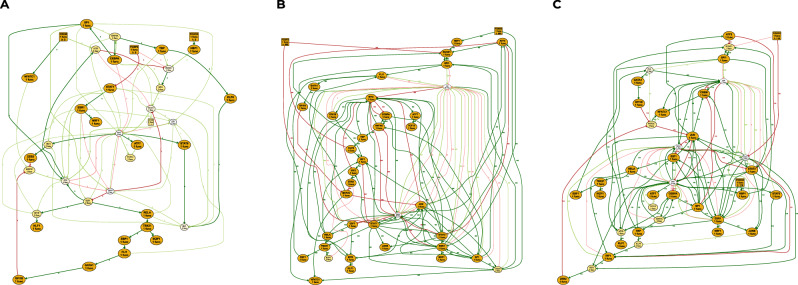
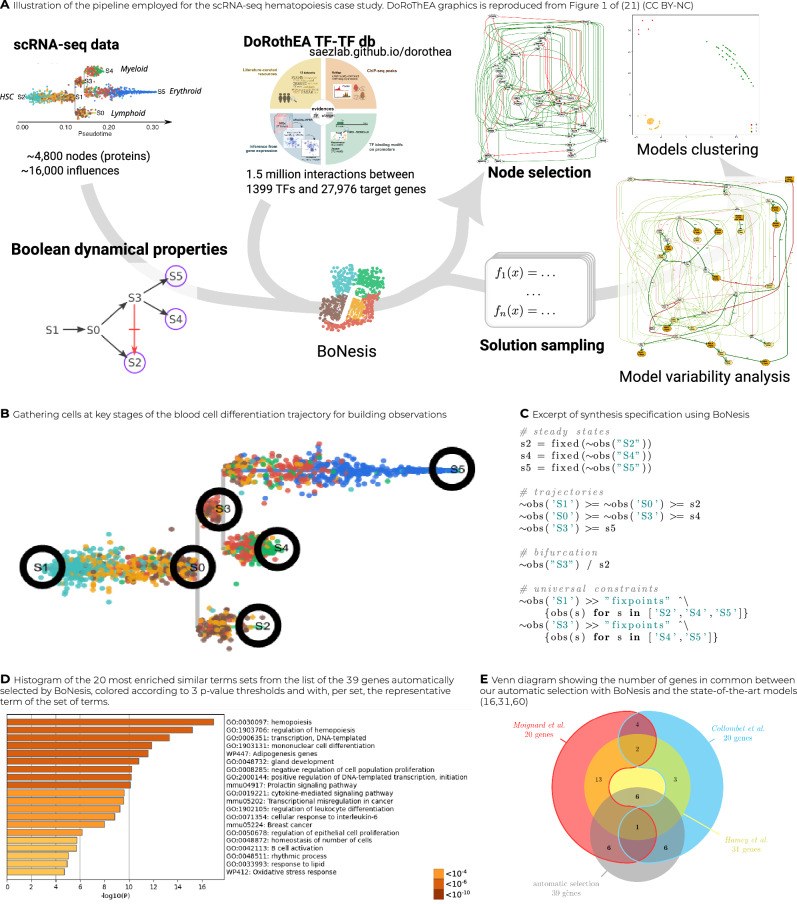
Boolean networks provide robust, explainable, and predictive models of cellular dynamics, especially for cellular differentiation and fate decision processes. Yet, the construction of such models is extremely challenging, as it requires integrating prior knowledge with experimental observation of the transcriptome, potentially relating thousands of genes. We present a general methodology for integrating transcriptome data and prior knowledge on the underlying gene regulatory network in order to generate automatically ensembles of Boolean networks able to reproduce the modeled qualitative behavior. Our methodology builds on the software BoNesis, which implements the automatic construction of Boolean networks from a specification of their expected structural and dynamical properties. We show how to transform transcriptome data into such a qualitative specification, and then how to exploit the generated ensembles of Boolean networks for identifying families of candidate models, and for predicting robust cellular reprogramming targets. We illustrate the scalability and versatility of our overall approach with two applications: the modeling of hematopoiesis from single-cell RNA-Seq data, and modeling the differentiation of bone marrow stromal cells into adipocytes and osteoblasts from bulk RNA-seq time series data. For this latter case, we took advantage of ensemble modeling to predict combinations of reprogramming factors for trans-differentiation that are robust to model uncertainties due to variations in experimental replicates and choice of binarization method. Moreover, we performed an in silico assessment of the fidelity and efficiency of the reprogramming and conducted preliminary experimental validation.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


