{"title":"Single-cell splicing QTL analysis in pancreatic islets.","authors":"Jae-Won Cho, Jingyi Cao, Martin Hemberg","doi":"10.3389/fbinf.2025.1657895","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Alternative splicing (AS) of mRNAs is a highly conserved mechanism which can greatly expand the functional diversity of the transcriptome. Aberrant splicing underpins many diseases, and a better understanding of AS can provide insights regarding the molecular mechanisms involved. Importantly, AS can be affected by genetic variants and several studies have indicated large numbers of splicing quantitative trait loci (sQTL). With the advance of single-cell technology, expression QTL studies have been expanded to identify cell type level variants.</p><p><strong>Methods: </strong>We collected eight full-length scRNA-seq pancreatic islet datasets. Genotyping for each individual was done by the CTAT pipeline and Streka2. The isoform quantification was done by RSEM. Finally, sQTL was obtained by sQTLseeker2.</p><p><strong>Results: </strong>As a result, we identified 228 cell type level sQTLs for alpha and beta cells across 152 genes. In particular, our study highlights four variants affecting CDC42, a gene related to cell morphology, which have not been observed from bulk sQTL analysis.</p><p><strong>Discussion: </strong>Our results provide a proof of concept that it is possible to identify cell type level sQTLs, and we envision that better powered studies will allow us to further uncover the genetic regulation of splicing.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"5 ","pages":"1657895"},"PeriodicalIF":3.9000,"publicationDate":"2025-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12457394/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2025.1657895","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Alternative splicing (AS) of mRNAs is a highly conserved mechanism which can greatly expand the functional diversity of the transcriptome. Aberrant splicing underpins many diseases, and a better understanding of AS can provide insights regarding the molecular mechanisms involved. Importantly, AS can be affected by genetic variants and several studies have indicated large numbers of splicing quantitative trait loci (sQTL). With the advance of single-cell technology, expression QTL studies have been expanded to identify cell type level variants.
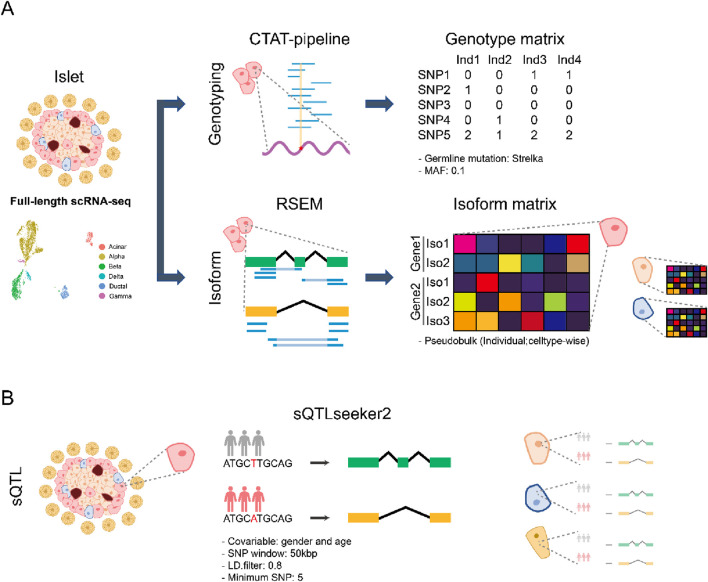
Methods: We collected eight full-length scRNA-seq pancreatic islet datasets. Genotyping for each individual was done by the CTAT pipeline and Streka2. The isoform quantification was done by RSEM. Finally, sQTL was obtained by sQTLseeker2.
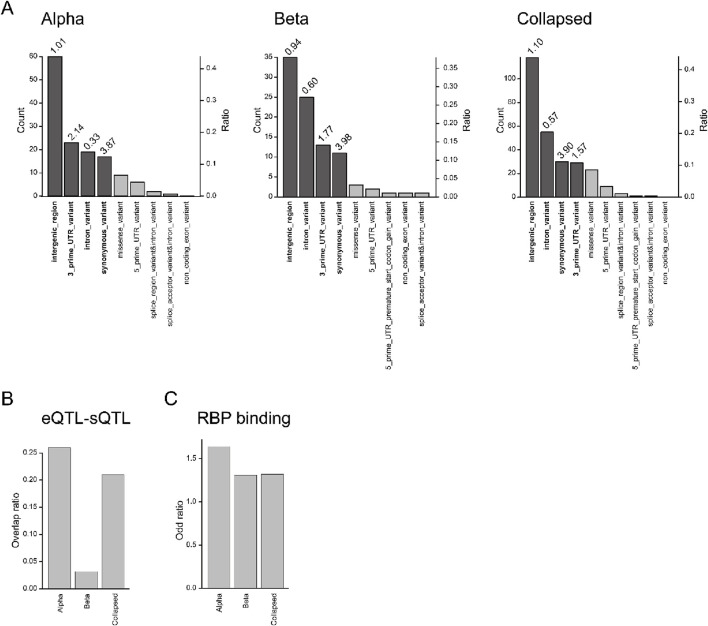
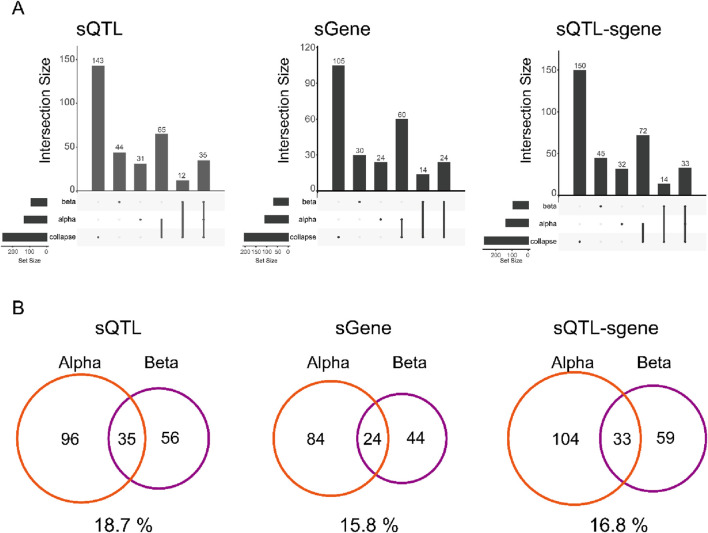
Results: As a result, we identified 228 cell type level sQTLs for alpha and beta cells across 152 genes. In particular, our study highlights four variants affecting CDC42, a gene related to cell morphology, which have not been observed from bulk sQTL analysis.
Discussion: Our results provide a proof of concept that it is possible to identify cell type level sQTLs, and we envision that better powered studies will allow us to further uncover the genetic regulation of splicing.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


