Genetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations
IF 2.4
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
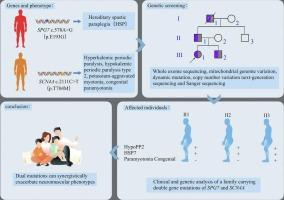
Hereditary spastic paraplegia (HSP) is a rare and genetically heterogeneous neurodegenerative disorder, primarily defined by progressive lower-limb spasticity and weakness. Among the numerous genes implicated, pathogenic variants in the spastic paraplegia 7 (SPG7) gene represent one of the most common causes of HSP, whereas mutations in SCN4A, a skeletal muscle ion channel gene, are typically associated with a diverse spectrum of phenotypes, including hyperkalemic and hypokalemic periodic paralysis, potassium-aggravated myotonia, and congenital paramyotonia. To date, however, the coexistence of pathogenic variants in SPG7 and SCN4A within the same pedigree, and their potential pathogenic interplay, has not been documented.
In this study, we performed comprehensive genetic profiling, including whole-exome sequencing, mitochondrial genome analysis, dynamic mutation screening, copy number variation assessment, and Sanger sequencing. We identified a novel heterozygous SPG7 variant (c.578A>G; p.E193G) alongside a known pathogenic SCN4A missense mutation (c.2111C>T; p.T704M). Remarkably, individuals harboring both variants presented with highly complex phenotypes that combined classical HSP manifestations with ion channel dysfunctions, such as congenital paramyotonia and hypokalemic periodic paralysis.
These findings provide the first evidence of a possible genetic interaction between SPG7 and SCN4A, expanding the recognized clinical and molecular spectrum of HSP. Our results underscore the diagnostic value of multi-gene testing in patients with atypical or overlapping neuromuscular symptoms and highlight the importance of considering potential polygenic contributions when interpreting the clinical heterogeneity of HSP.
SCN4A和SPG7双突变致骨骼肌离子通道病和遗传性痉挛性截瘫7型家族遗传分析
遗传性痉挛性截瘫(HSP)是一种罕见的遗传异质性神经退行性疾病,主要表现为进行性下肢痉挛和无力。在涉及的众多基因中,痉挛性截瘫7 (SPG7)基因的致病性变异是HSP的最常见原因之一,而骨骼肌离子通道基因SCN4A的突变通常与多种表型相关,包括高钾血症和低钾血症周期性麻痹、钾加剧型肌强直和先天性肌张力异常。然而,迄今为止,SPG7和SCN4A的致病变异在同一家系中共存,以及它们潜在的致病相互作用尚未得到证实。在这项研究中,我们进行了全面的遗传分析,包括全外显子组测序、线粒体基因组分析、动态突变筛选、拷贝数变异评估和Sanger测序。我们发现了一种新的杂合SPG7变异(c.578A>G; p.E193G)和一种已知的致病性SCN4A错义突变(c.2111C>T; p.T704M)。值得注意的是,携带这两种变体的个体表现出高度复杂的表型,将经典的热休克表现与离子通道功能障碍相结合,如先天性肌张力异常和低钾性周期性麻痹。这些发现为SPG7和SCN4A之间可能的遗传相互作用提供了第一个证据,扩大了公认的热休克蛋白的临床和分子谱。我们的研究结果强调了多基因检测在非典型或重叠神经肌肉症状患者中的诊断价值,并强调了在解释热休克的临床异质性时考虑潜在多基因贡献的重要性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Gene
生物-遗传学
CiteScore
6.10
自引率
2.90%
发文量
718
审稿时长
42 days
期刊介绍:
Gene publishes papers that focus on the regulation, expression, function and evolution of genes in all biological contexts, including all prokaryotic and eukaryotic organisms, as well as viruses.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


