Antoine Dubray-Vautrin, Victor Gravrand, Grégoire Marret, Constance Lamy, Jerzy Klijanienko, Sophie Vacher, Ladidi Ahmanache, Maud Kamal, Olivier Choussy, Nicolas Servant, Célia Dupain, Christophe Le Tourneau, Jimmy Mullaert
{"title":"Internal validation strategy for high dimensional prognosis model: A simulation study and application to transcriptomic in head and neck tumors.","authors":"Antoine Dubray-Vautrin, Victor Gravrand, Grégoire Marret, Constance Lamy, Jerzy Klijanienko, Sophie Vacher, Ladidi Ahmanache, Maud Kamal, Olivier Choussy, Nicolas Servant, Célia Dupain, Christophe Le Tourneau, Jimmy Mullaert","doi":"10.1016/j.csbj.2025.08.035","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Predictive models using high-dimensional data, such as genomics and transcriptomics, are increasingly used in oncology for time-to-event endpoints. Internal validation of these models is crucial to mitigate optimism bias prior to external validation. Common strategies include train-test, bootstrap, and (nested) cross-validation. However, no benchmark exists for these methods in high-dimensional settings. We aimed to compare these strategies and provide recommendations in the field of transcriptomic analysis.</p><p><strong>Method: </strong>A simulation study was conducted using data from the SCANDARE head and neck cohort (NCT03017573) including n = 76 patients. Simulated datasets included clinical variables (age, sex, HPV status, TNM staging), transcriptomic data (15,000 transcripts), and disease-free survival, with a realistic cumulative baseline hazard. Sample sizes of 50, 75, 100, 500, and 1000 were simulated, with 100 replicates each. Cox penalized regression was performed for model selection, followed by train-test 70 % training), bootstrap (100 iterations), 5-fold cross-validation, and nested cross-validation (5 ×5) to assess discriminative (time-dependent AUC and C-Index) and calibration (3-year integrated Brier Score) performance.</p><p><strong>Results: </strong>Train-test validation showed unstable performance. Conventional bootstrap was over-optimistic, while the 0.632 + bootstrap was overly pessimistic, particularly with small samples (n = 50 to n = 100). The k-fold cross-validation and nested cross-validation improved performance with larger sample sizes, with k-fold cross-validation demonstrating greater stability. Nested cross-validation showed performance fluctuations depending on the regularization method for model development.</p><p><strong>Conclusion: </strong>The K-fold cross-validation and nested cross-validation are recommended for internal validation of Cox penalized models in high-dimensional time-to-event settings. These methods offer greater stability and reliability compared to train-test or bootstrap approaches, particularly when sample sizes are sufficient.</p>","PeriodicalId":10715,"journal":{"name":"Computational and structural biotechnology journal","volume":"27 ","pages":"3792-3802"},"PeriodicalIF":4.1000,"publicationDate":"2025-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12451366/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and structural biotechnology journal","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.csbj.2025.08.035","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Predictive models using high-dimensional data, such as genomics and transcriptomics, are increasingly used in oncology for time-to-event endpoints. Internal validation of these models is crucial to mitigate optimism bias prior to external validation. Common strategies include train-test, bootstrap, and (nested) cross-validation. However, no benchmark exists for these methods in high-dimensional settings. We aimed to compare these strategies and provide recommendations in the field of transcriptomic analysis.
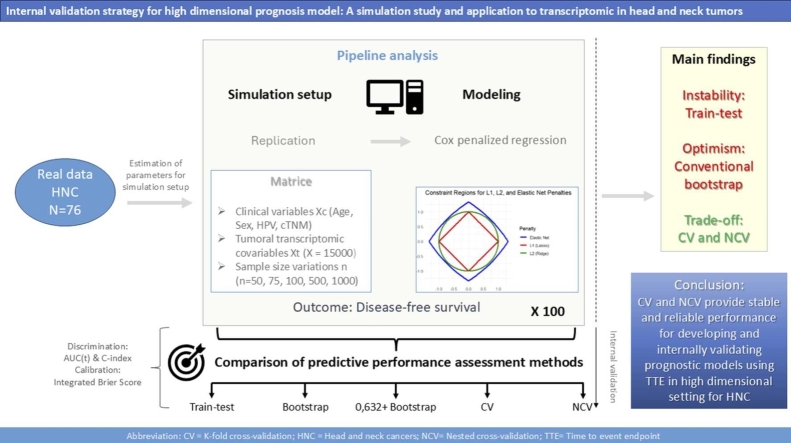
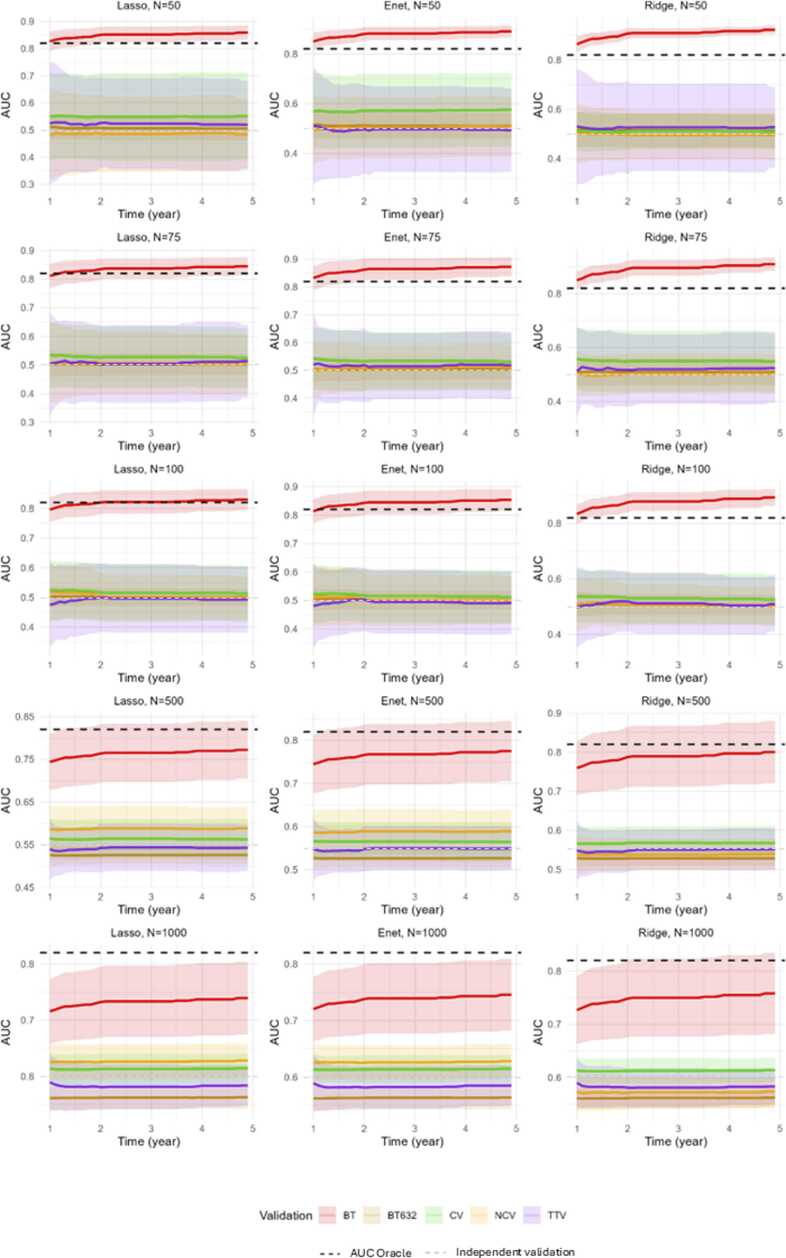
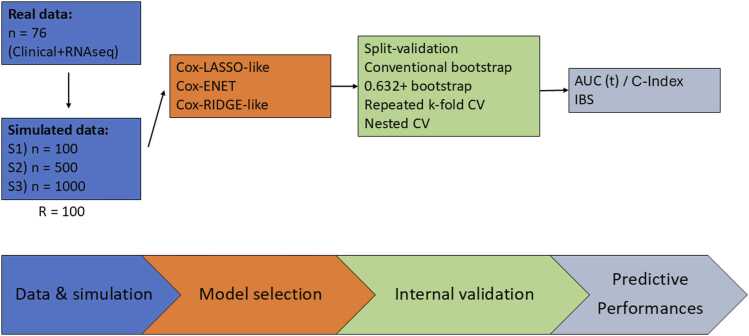
Method: A simulation study was conducted using data from the SCANDARE head and neck cohort (NCT03017573) including n = 76 patients. Simulated datasets included clinical variables (age, sex, HPV status, TNM staging), transcriptomic data (15,000 transcripts), and disease-free survival, with a realistic cumulative baseline hazard. Sample sizes of 50, 75, 100, 500, and 1000 were simulated, with 100 replicates each. Cox penalized regression was performed for model selection, followed by train-test 70 % training), bootstrap (100 iterations), 5-fold cross-validation, and nested cross-validation (5 ×5) to assess discriminative (time-dependent AUC and C-Index) and calibration (3-year integrated Brier Score) performance.
Results: Train-test validation showed unstable performance. Conventional bootstrap was over-optimistic, while the 0.632 + bootstrap was overly pessimistic, particularly with small samples (n = 50 to n = 100). The k-fold cross-validation and nested cross-validation improved performance with larger sample sizes, with k-fold cross-validation demonstrating greater stability. Nested cross-validation showed performance fluctuations depending on the regularization method for model development.
Conclusion: The K-fold cross-validation and nested cross-validation are recommended for internal validation of Cox penalized models in high-dimensional time-to-event settings. These methods offer greater stability and reliability compared to train-test or bootstrap approaches, particularly when sample sizes are sufficient.
期刊介绍:
Computational and Structural Biotechnology Journal (CSBJ) is an online gold open access journal publishing research articles and reviews after full peer review. All articles are published, without barriers to access, immediately upon acceptance. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together through the application of computational methods. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal. Specific areas of interest include, but are not limited to:
Structure and function of proteins, nucleic acids and other macromolecules
Structure and function of multi-component complexes
Protein folding, processing and degradation
Enzymology
Computational and structural studies of plant systems
Microbial Informatics
Genomics
Proteomics
Metabolomics
Algorithms and Hypothesis in Bioinformatics
Mathematical and Theoretical Biology
Computational Chemistry and Drug Discovery
Microscopy and Molecular Imaging
Nanotechnology
Systems and Synthetic Biology

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


