David Ferreiro, Luis Daniel González-Vázquez, Ana Prado-Comesaña, Miguel Arenas
{"title":"Forecasting protein evolution by integrating birth-death population models with structurally constrained substitution models.","authors":"David Ferreiro, Luis Daniel González-Vázquez, Ana Prado-Comesaña, Miguel Arenas","doi":"10.7554/eLife.106365","DOIUrl":null,"url":null,"abstract":"<p><p>Evolutionary studies in population genetics and ecology were mainly focused on predicting and understanding past evolutionary events. Recently, however, a growing trend explores the prediction of evolutionary trajectories toward the future promoted by its wide variety of applications. In this context, we introduce a forecasting protein evolution method that integrates birth-death population models with substitution models that consider selection on protein folding stability. In contrast to traditional population genetics methods that usually make the unrealistic assumption of simulating molecular evolution separately from the evolutionary history, the present method combines both processes to simultaneously model forward-in-time birth-death evolutionary trajectories and protein evolution under structurally constrained substitution models that outperformed traditional empirical substitution models. We implemented the method into a freely available computer framework. We evaluated the accuracy of the predictions with several monitored viral proteins of broad interest. Overall, the method showed acceptable errors in predicting the folding stability of the forecasted protein variants, but, expectedly, the errors were larger in the prediction of the corresponding sequences. We conclude that forecasting protein evolution is feasible in certain evolutionary scenarios and provide suggestions to enhance its accuracy by improving the underlying models of evolution.</p>","PeriodicalId":11640,"journal":{"name":"eLife","volume":"14 ","pages":""},"PeriodicalIF":6.4000,"publicationDate":"2025-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12459951/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"eLife","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.7554/eLife.106365","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
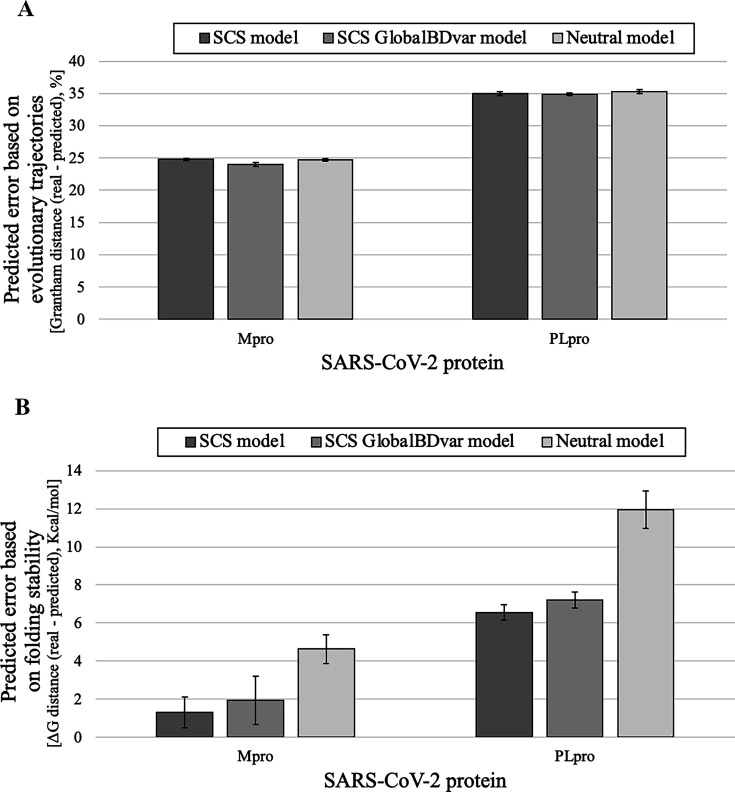
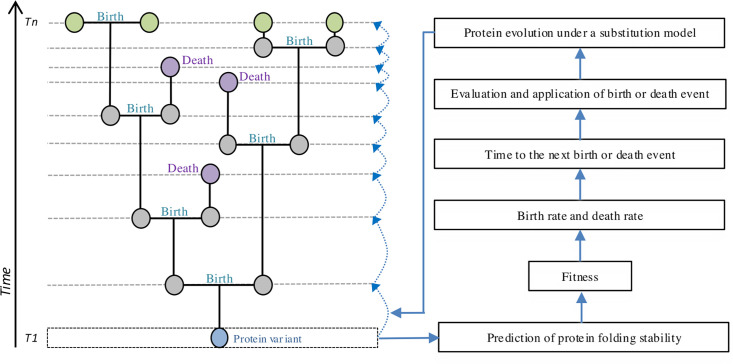
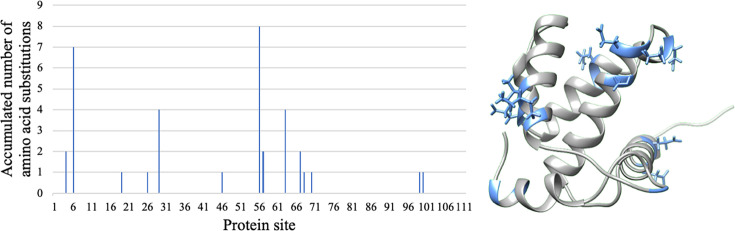
Evolutionary studies in population genetics and ecology were mainly focused on predicting and understanding past evolutionary events. Recently, however, a growing trend explores the prediction of evolutionary trajectories toward the future promoted by its wide variety of applications. In this context, we introduce a forecasting protein evolution method that integrates birth-death population models with substitution models that consider selection on protein folding stability. In contrast to traditional population genetics methods that usually make the unrealistic assumption of simulating molecular evolution separately from the evolutionary history, the present method combines both processes to simultaneously model forward-in-time birth-death evolutionary trajectories and protein evolution under structurally constrained substitution models that outperformed traditional empirical substitution models. We implemented the method into a freely available computer framework. We evaluated the accuracy of the predictions with several monitored viral proteins of broad interest. Overall, the method showed acceptable errors in predicting the folding stability of the forecasted protein variants, but, expectedly, the errors were larger in the prediction of the corresponding sequences. We conclude that forecasting protein evolution is feasible in certain evolutionary scenarios and provide suggestions to enhance its accuracy by improving the underlying models of evolution.
期刊介绍:
eLife is a distinguished, not-for-profit, peer-reviewed open access scientific journal that specializes in the fields of biomedical and life sciences. eLife is known for its selective publication process, which includes a variety of article types such as:
Research Articles: Detailed reports of original research findings.
Short Reports: Concise presentations of significant findings that do not warrant a full-length research article.
Tools and Resources: Descriptions of new tools, technologies, or resources that facilitate scientific research.
Research Advances: Brief reports on significant scientific advancements that have immediate implications for the field.
Scientific Correspondence: Short communications that comment on or provide additional information related to published articles.
Review Articles: Comprehensive overviews of a specific topic or field within the life sciences.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


