Copper overload worsens the inflammatory response of microglia to amyloid beta (Aβ) by impairing phagocytosis and promoting mitochondrial DNA-mediated NLRP3 inflammasome activation
IF 11.9
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Microglia play a significant role in the development and progression of Alzheimer's disease (AD). These brain-resident immune cells efficiently clear neurotoxic amyloid beta (Aβ) peptides; however, chronic activation may overwhelm their protective abilities, resulting in persistent neuroinflammation. The causes of aberrant microglial activation in AD remain elusive. Emerging evidence indicates that copper (Cu) accumulation, which can arise from prolonged exposure to various environmental sources, modifies the innate immune response in AD. Here, we sought to explore the mechanisms by which Cu overload regulates the microglial phenotype when exposed to Aβ. Our findings showed that exposure to sublethal doses of Cu led to the accumulation of this transition metal in the mitochondria. Elevated mitochondrial Cu (mtCu) levels were accompanied by reduced mitochondrial glutathione (mtGSH) and high oxidative stress, leading to Aβ-induced inflammasome activation through the release of oxidized mitochondrial DNA (ox-mtDNA). Moreover, increased intracellular Cu levels enhanced cholesterol biosynthesis and facilitated its transport to mitochondria. The combination of elevated cholesterol and mitochondrial oxidative stress hindered the ability of microglia to phagocytose Aβ effectively. As expected, conditioned medium from Cu-activated microglia reduced neuronal viability. The neurotoxicity caused by Cu-overloaded microglia was prevented by inhibiting inflammasome activation and restoring mtGSH levels. In conclusion, our study outlines a mechanistic pathway by which chronic exposure to environmental Cu may lead to neuroinflammation and Aβ accumulation in AD, underscoring the crucial role of mitochondrial oxidative stress.
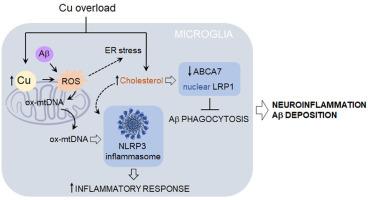
铜超载通过损害吞噬和促进线粒体dna介导的NLRP3炎性小体激活,加重了小胶质细胞对β -淀粉样蛋白(Aβ)的炎症反应
小胶质细胞在阿尔茨海默病(AD)的发生和发展中起着重要作用。这些脑内免疫细胞有效清除神经毒性淀粉样蛋白(Aβ)肽;然而,慢性激活可能会压倒它们的保护能力,导致持续的神经炎症。阿尔茨海默氏症小胶质细胞异常激活的原因尚不清楚。新出现的证据表明,铜(Cu)的积累,可引起长期暴露于各种环境来源,改变先天免疫反应的阿尔茨海默病。在这里,我们试图探索当暴露于Aβ时,Cu过载调节小胶质细胞表型的机制。我们的研究结果表明,暴露于亚致死剂量的铜导致线粒体中这种过渡金属的积累。线粒体Cu (mtCu)水平升高伴随着线粒体谷胱甘肽(mtGSH)减少和高氧化应激,通过释放氧化线粒体DNA (ox-mtDNA)导致a β诱导的炎性体活化。此外,细胞内Cu水平的增加促进了胆固醇的生物合成并促进了其向线粒体的运输。升高的胆固醇和线粒体氧化应激的结合阻碍了小胶质细胞有效吞噬Aβ的能力。正如预期的那样,来自cu激活的小胶质细胞的条件培养基降低了神经元的活力。通过抑制炎性体激活和恢复mtGSH水平,可以预防cu超载小胶质细胞引起的神经毒性。总之,我们的研究概述了慢性暴露于环境Cu可能导致AD神经炎症和a β积累的机制途径,强调了线粒体氧化应激的关键作用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Redox Biology
BIOCHEMISTRY & MOLECULAR BIOLOGY-
CiteScore
19.90
自引率
3.50%
发文量
318
审稿时长
25 days
期刊介绍:
Redox Biology is the official journal of the Society for Redox Biology and Medicine and the Society for Free Radical Research-Europe. It is also affiliated with the International Society for Free Radical Research (SFRRI). This journal serves as a platform for publishing pioneering research, innovative methods, and comprehensive review articles in the field of redox biology, encompassing both health and disease.
Redox Biology welcomes various forms of contributions, including research articles (short or full communications), methods, mini-reviews, and commentaries. Through its diverse range of published content, Redox Biology aims to foster advancements and insights in the understanding of redox biology and its implications.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


