{"title":"Recombination and phylogenetic inference.","authors":"Bruce Rannala","doi":"10.1093/evolinnean/kzaf016","DOIUrl":null,"url":null,"abstract":"<p><p>I explore the problem of inferring phylogenetic trees in the presence of recombination. Two widely used approaches are considered: concatenation methods assume all loci have one underlying gene tree; and species tree inference methods assume one gene tree underlies each locus (no intralocus recombination) and loci have independent gene trees (high interlocus recombination). The impact of recombination is different under these two approaches. Three strategies for addressing the impacts of recombination are considered: (i) studies of the statistical robustness of phylogenetic inference methods when recombination occurs and is not accounted for (if impacts are minimal, recombination can be safely ignored); (ii) methods that accommodate recombination by identifying recombinant regions to either eliminate recombinant loci (to reduce intralocus recombination) or to choose loci that are separated by multiple recombinations (to increase interlocus recombination); and (iii) methods for phylogenetic inference that aim to accommodate recombination by inferring breakpoints between regions of sequences with different gene trees or allow varying topology along a sequence. I conclude that recombination is likely to be more detrimental for concatenation methods, having little impact on topology or divergence time estimates for species tree inference methods. Recombination detection may not be necessary when performing species tree inference, and eliminating recombinant loci may bias parameter estimates. Methods allowing gene trees to vary across the genome still lack theory-based criteria for combining inferred gene trees to estimate a species tree; this could in principle be done using a multispecies coalescent model with recombination but is a considerable technical challenge.</p>","PeriodicalId":520301,"journal":{"name":"Evolutionary journal of the Linnean Society","volume":"4 1","pages":"kzaf016"},"PeriodicalIF":0.0000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12448323/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary journal of the Linnean Society","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/evolinnean/kzaf016","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
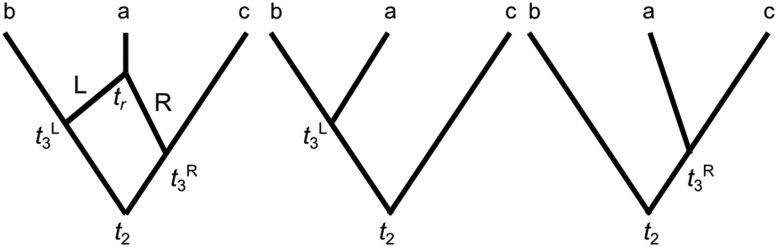
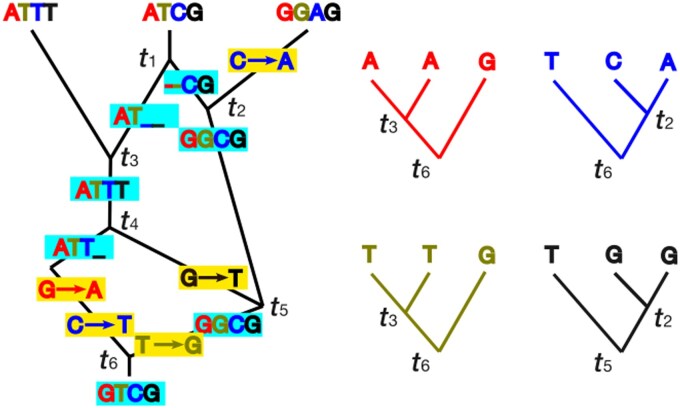
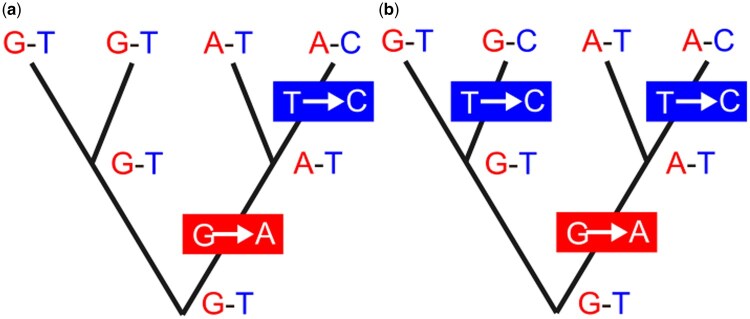
I explore the problem of inferring phylogenetic trees in the presence of recombination. Two widely used approaches are considered: concatenation methods assume all loci have one underlying gene tree; and species tree inference methods assume one gene tree underlies each locus (no intralocus recombination) and loci have independent gene trees (high interlocus recombination). The impact of recombination is different under these two approaches. Three strategies for addressing the impacts of recombination are considered: (i) studies of the statistical robustness of phylogenetic inference methods when recombination occurs and is not accounted for (if impacts are minimal, recombination can be safely ignored); (ii) methods that accommodate recombination by identifying recombinant regions to either eliminate recombinant loci (to reduce intralocus recombination) or to choose loci that are separated by multiple recombinations (to increase interlocus recombination); and (iii) methods for phylogenetic inference that aim to accommodate recombination by inferring breakpoints between regions of sequences with different gene trees or allow varying topology along a sequence. I conclude that recombination is likely to be more detrimental for concatenation methods, having little impact on topology or divergence time estimates for species tree inference methods. Recombination detection may not be necessary when performing species tree inference, and eliminating recombinant loci may bias parameter estimates. Methods allowing gene trees to vary across the genome still lack theory-based criteria for combining inferred gene trees to estimate a species tree; this could in principle be done using a multispecies coalescent model with recombination but is a considerable technical challenge.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


