Irene Medina-Martínez, Rocío Gil-Gutiérrez, Jorge García-García, Francisco Javier de la Hera-Fernández, Nuria Navarrete-Navarrete, Mónica Zamora-Pasadas, Norberto Ortego-Centeno, José Luis Callejas-Rubio, Federico García-García, Julio Gálvez-Peralta, Alba Rodríguez-Nogales, María Correa-Rodríguez, Blanca Rueda-Medina
{"title":"Association of Gut Dysbiosis with Disease Phenotype and Treatment in Systemic Lupus Erythematosus.","authors":"Irene Medina-Martínez, Rocío Gil-Gutiérrez, Jorge García-García, Francisco Javier de la Hera-Fernández, Nuria Navarrete-Navarrete, Mónica Zamora-Pasadas, Norberto Ortego-Centeno, José Luis Callejas-Rubio, Federico García-García, Julio Gálvez-Peralta, Alba Rodríguez-Nogales, María Correa-Rodríguez, Blanca Rueda-Medina","doi":"10.3390/medsci13030151","DOIUrl":null,"url":null,"abstract":"<p><p><b>Introduction</b>: Gut dysbiosis has been associated with the development of autoimmune diseases, including systemic lupus erythematosus (SLE). Although previous studies suggest microbial alterations in SLE, evidence at the species level and its clinical relevance remain limited. This study aimed to characterise the gut microbiota at species level in SLE patients and evaluate its association with clinical features. <b>Materials and methods</b>: A total of 57 SLE patients and 57 matched controls were included. Faecal samples were collected using the OMNIgene-GUT kit, and microbial DNA was extracted with the Maxwell RSC PureFood GMO kit. Metagenomic sequencing was performed using the Illumina MiSeq platform, and the data was analysed with QIIME2. Microbial diversity and relative abundance were assessed using the phyloseq package, and differentially abundant taxa were identified using DESeq2. Clinical subgroups among SLE patients were identified via k-means clustering. <b>Results</b>: SLE patients exhibited significantly different beta diversity compared to controls (<i>p</i> = 0.001), with increased abundance of <i>Pseudomonadota</i> (3.81% vs. 6.80%, <i>p</i> < 0.05) and decreased <i>Bacteroidota</i> (53.42% vs. 38.04%, <i>p</i> < 0.05). Only 10 bacterial species were consistently present across all SLE samples, including <i>Akkermansia muciniphila</i>, <i>Bacteroides dorei</i>, and <i>Lactobacillus gasseri.</i> Hypertensive patients and those treated with corticosteroids presented a marked depletion of key microbial taxa. Conversely, Belimumab-treated patients displayed a distinct microbiota enriched in species such as <i>Alistipes shahii</i> and <i>Prevotella corporis</i>. <b>Conclusions</b>: This study confirms significant gut microbiota alterations in SLE and pinpoints microbial profiles associated with clinical subgroups. These findings suggest gut dysbiosis may contribute to SLE pathogenesis and indicate biomarkers for disease stratification.</p>","PeriodicalId":74152,"journal":{"name":"Medical sciences (Basel, Switzerland)","volume":"13 3","pages":""},"PeriodicalIF":4.4000,"publicationDate":"2025-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12452298/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medical sciences (Basel, Switzerland)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/medsci13030151","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
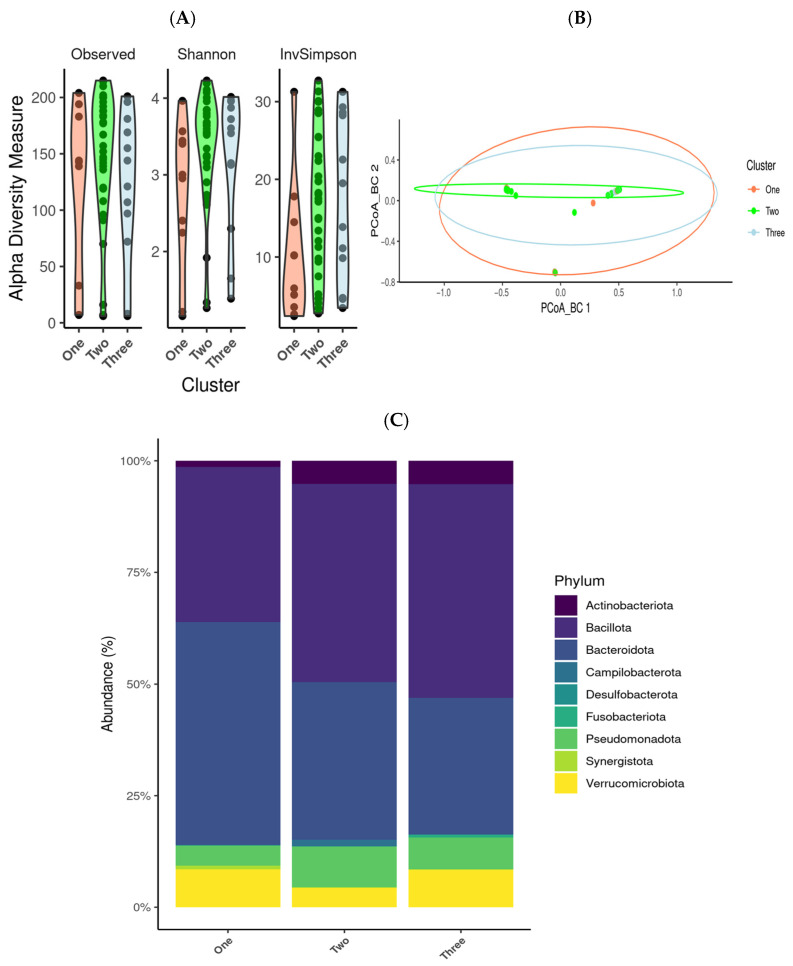
Introduction: Gut dysbiosis has been associated with the development of autoimmune diseases, including systemic lupus erythematosus (SLE). Although previous studies suggest microbial alterations in SLE, evidence at the species level and its clinical relevance remain limited. This study aimed to characterise the gut microbiota at species level in SLE patients and evaluate its association with clinical features. Materials and methods: A total of 57 SLE patients and 57 matched controls were included. Faecal samples were collected using the OMNIgene-GUT kit, and microbial DNA was extracted with the Maxwell RSC PureFood GMO kit. Metagenomic sequencing was performed using the Illumina MiSeq platform, and the data was analysed with QIIME2. Microbial diversity and relative abundance were assessed using the phyloseq package, and differentially abundant taxa were identified using DESeq2. Clinical subgroups among SLE patients were identified via k-means clustering. Results: SLE patients exhibited significantly different beta diversity compared to controls (p = 0.001), with increased abundance of Pseudomonadota (3.81% vs. 6.80%, p < 0.05) and decreased Bacteroidota (53.42% vs. 38.04%, p < 0.05). Only 10 bacterial species were consistently present across all SLE samples, including Akkermansia muciniphila, Bacteroides dorei, and Lactobacillus gasseri. Hypertensive patients and those treated with corticosteroids presented a marked depletion of key microbial taxa. Conversely, Belimumab-treated patients displayed a distinct microbiota enriched in species such as Alistipes shahii and Prevotella corporis. Conclusions: This study confirms significant gut microbiota alterations in SLE and pinpoints microbial profiles associated with clinical subgroups. These findings suggest gut dysbiosis may contribute to SLE pathogenesis and indicate biomarkers for disease stratification.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


