{"title":"MADVAR: a lightweight, data-driven tool for automated feature selection in omics data.","authors":"Gilad Silberberg","doi":"10.1093/bioadv/vbaf211","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>High-throughput biological data provides rich opportunities for discovery, but its vastness leads to the inclusion of many irrelevant features that hinder effective analysis, especially in unsupervised clustering and machine learning tasks. Traditional feature selection methods such as correlation filtering, PCA, mutual information, and Laplacian scores often either eliminate important features or demand extensive computational resources, and their thresholds are usually arbitrary rather than data-driven.</p><p><strong>Results: </strong>MADVAR addresses these challenges as a lightweight R package for automated feature selection in omics data, introducing two data-driven methods-madvar and intersectDistributions-that define thresholds based on the statistical structure of the data itself. These approaches eliminate the reliance on arbitrary cutoffs and efficiently filter features without expensive computation. Benchmarking across diverse omics datasets shows that MADVAR achieves top performance in clustering and classification tasks while maintaining computational efficiency, and it integrates seamlessly into existing R-based analysis pipelines.</p><p><strong>Availability and implementation: </strong>The source code and documentation for MADVAR are freely available on GitHub (https://github.com/Champions-Oncology/MADVAR). The package is implemented in R and runs on all major operating systems.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf211"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12449246/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf211","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: High-throughput biological data provides rich opportunities for discovery, but its vastness leads to the inclusion of many irrelevant features that hinder effective analysis, especially in unsupervised clustering and machine learning tasks. Traditional feature selection methods such as correlation filtering, PCA, mutual information, and Laplacian scores often either eliminate important features or demand extensive computational resources, and their thresholds are usually arbitrary rather than data-driven.
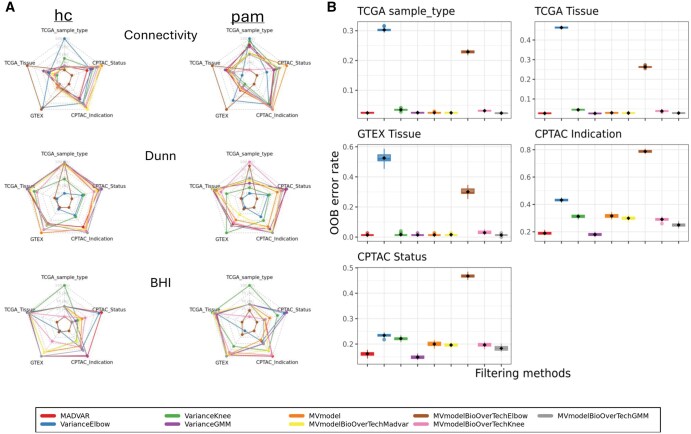
Results: MADVAR addresses these challenges as a lightweight R package for automated feature selection in omics data, introducing two data-driven methods-madvar and intersectDistributions-that define thresholds based on the statistical structure of the data itself. These approaches eliminate the reliance on arbitrary cutoffs and efficiently filter features without expensive computation. Benchmarking across diverse omics datasets shows that MADVAR achieves top performance in clustering and classification tasks while maintaining computational efficiency, and it integrates seamlessly into existing R-based analysis pipelines.
Availability and implementation: The source code and documentation for MADVAR are freely available on GitHub (https://github.com/Champions-Oncology/MADVAR). The package is implemented in R and runs on all major operating systems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


