Ilja Dubinski, Belana Debor, Sofia Petrova, Katharina A Schiergens, Heike Weigand, Heinrich Schmidt
{"title":"MCT8 Deficiency in Infancy: Opportunities for Early Diagnosis and Screening.","authors":"Ilja Dubinski, Belana Debor, Sofia Petrova, Katharina A Schiergens, Heike Weigand, Heinrich Schmidt","doi":"10.3390/ijns11030066","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Monocarboxylate-transporter-8-(MCT8) deficiency, or Allan-Herndon-Dudley syndrome (AHDS), is a rare X-linked disorder caused by pathogenic variants in the SLC16A2 gene, leading to impaired transport of thyroid hormones, primarily T3 and T4, across cell membranes. The resulting central hypothyroidism and peripheral hyperthyroidism cause neurodevelopmental impairment and thyrotoxicosis. Despite the availability of therapy options, e.g., with triiodothyroacetic acid (TRIAC), diagnosis is often delayed, partly due to normal TSH levels or incomplete genetic panels. MCT8 deficiency is not yet included in newborn-screening programs worldwide.</p><p><strong>Case description: </strong>We present a case of an infant genetically diagnosed with MCT8 deficiency at 5 months of age after presenting with muscular hypotonia, lack of head control, and developmental delay. Thyroid function testing revealed a normal TSH, low free T4, and significantly elevated free T3 and free T3/T4 ratio. Treatment with TRIAC (Emcitate<sup>®</sup>) was initiated promptly, with close drug monitoring. Despite persistent motor deficits and dystonia, some developmental progress was observed, as well as reduction in hyperthyroidism.</p><p><strong>Discussion/conclusions: </strong>This case underscores the importance of early free T3 and fT3/fT4 ratio testing in infants with unexplained developmental delay. Broader inclusion of SLC16A2 in genetic panels and consideration of newborn screening could improve early diagnosis and outcomes in this rare but treatable condition.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 3","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12452532/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11030066","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Monocarboxylate-transporter-8-(MCT8) deficiency, or Allan-Herndon-Dudley syndrome (AHDS), is a rare X-linked disorder caused by pathogenic variants in the SLC16A2 gene, leading to impaired transport of thyroid hormones, primarily T3 and T4, across cell membranes. The resulting central hypothyroidism and peripheral hyperthyroidism cause neurodevelopmental impairment and thyrotoxicosis. Despite the availability of therapy options, e.g., with triiodothyroacetic acid (TRIAC), diagnosis is often delayed, partly due to normal TSH levels or incomplete genetic panels. MCT8 deficiency is not yet included in newborn-screening programs worldwide.
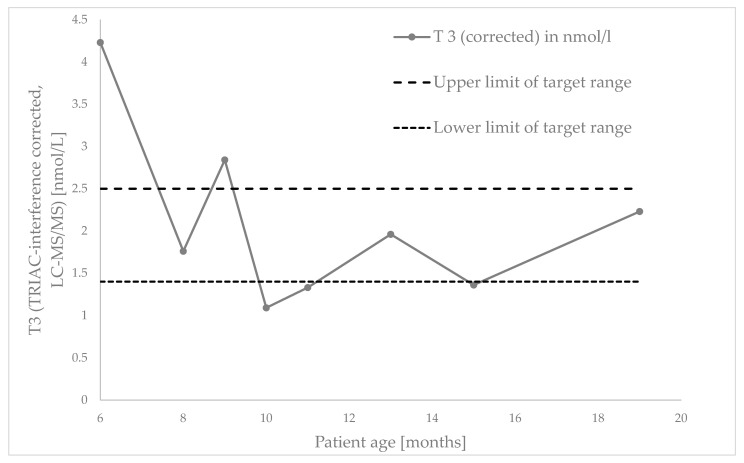
Case description: We present a case of an infant genetically diagnosed with MCT8 deficiency at 5 months of age after presenting with muscular hypotonia, lack of head control, and developmental delay. Thyroid function testing revealed a normal TSH, low free T4, and significantly elevated free T3 and free T3/T4 ratio. Treatment with TRIAC (Emcitate®) was initiated promptly, with close drug monitoring. Despite persistent motor deficits and dystonia, some developmental progress was observed, as well as reduction in hyperthyroidism.
Discussion/conclusions: This case underscores the importance of early free T3 and fT3/fT4 ratio testing in infants with unexplained developmental delay. Broader inclusion of SLC16A2 in genetic panels and consideration of newborn screening could improve early diagnosis and outcomes in this rare but treatable condition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


