Structure search for B7Mn2 clusters: Inverse sandwich geometry with a high-spin state
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Herein, we present a density functional theory (DFT) investigation of the BMn cluster, a boron-based system doped with two manganese atoms. The most stable structure adopts an inverse sandwich configuration, in which the B ring is symmetrically coordinated by two Mn atoms and exhibits a spin multiplicity of eight. Higher-energy isomers retain the B wheel-like motif, with Mn atoms positioned either above the ring or at peripheral sites. The Mn–B complex exhibits moderate interaction energy, arising from a balance between favorable electrostatic and orbital contributions and significant Pauli repulsion. Strong -type interactions between the Mn -orbitals and the delocalized B ring lead to substantial charge transfer (), rendering the Mn centers electron-deficient. This behavior is consistent with their Lewis acidic character and a weak Mn–Mn bonding interaction. Nucleus-independent chemical shift (NICS) isosurface analysis reveals a pronounced antiaromatic character, with extended deshielding under a magnetic field applied along the -axis. In contrast, fields oriented along the - and -directions produce more localized effects, highlighting the planar delocalization of the antiaromatic B framework.
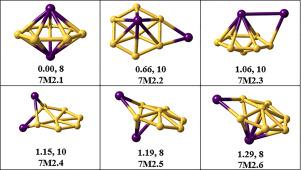
B7Mn2簇的结构搜索:具有高自旋态的逆夹心几何
在这里,我们提出了密度泛函理论(DFT)研究B7Mn2簇,一个掺杂两个锰原子的硼基体系。最稳定的结构是逆夹心结构,其中B7环由两个Mn原子对称配位,自旋多重数为8。高能异构体保留了B7轮状基序,Mn原子位于环的上方或外围位置。Mn2-B7配合物表现出适度的相互作用能,这是由于有利的静电和轨道贡献以及显著的泡利排斥之间的平衡。Mn -d轨道与离域B7环之间的强π型相互作用导致大量电荷转移(~ 1.3e−),导致Mn中心缺电子。这与它们的刘易斯酸性和弱的Mn-Mn键相互作用是一致的。核无关化学位移(NICS)等值面分析显示出明显的反芳特性,在沿z轴施加的磁场下具有扩展的去屏蔽。相反,沿x和y方向取向的电场产生更多的局部效应,突出了反芳B7框架的平面非局部化。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


