Ricci-GraphDTA: A graph neural network integrating discrete Ricci curvature for drug–target affinity prediction
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Drug–target affinity (DTA) prediction facilitates accelerated drug screening and reduces development costs. To enhance prediction performance and generalization capability, this paper proposes a DTA prediction model based on discrete curvature, named Ricci-GraphDTA, which integrates molecular graph and protein sequence modeling for efficient and accurate DTA prediction. The model consists of three parts: feature encoding, input representation learning, and affinity prediction. In the feature encoding stage, drug molecules are modeled as graphs, where Forman curvature is introduced to adjust the weights of neighbor information aggregation. A GIN residual network is then used to capture the local geometric and topological features of molecules. Protein sequences are modeled using BiLSTM to extract global dependency features, enhanced by an attention mechanism to capture long-range dependencies and key residue interactions—overcoming the limitations of traditional CNNs in handling long-range dependencies. In the input representation learning stage, the high-level representations of drugs and proteins are concatenated and passed through multiple nonlinear transformations to extract cross-modal interaction features, which are then used for affinity prediction. Experimental results demonstrate that Ricci-GraphDTA exhibits significant performance across various evaluation metrics on the Davis and KIBA datasets. Further cold-start experiments demonstrate the strong generalization ability of Ricci-GraphDTA in scenarios involving unseen drugs or targets, highlighting its potential in real-world drug discovery applications. On average, it achieves a 22.5% reduction in MSE across three cold-start tasks, with over 42% reduction in the dual cold-start setting, showcasing excellent structural modeling capability and robustness.
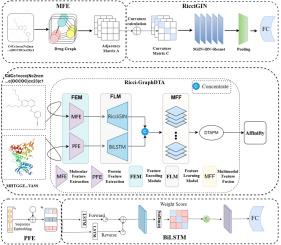
Ricci- graphdta:一种集成离散Ricci曲率的图神经网络,用于药物靶点亲和力预测。
药物靶标亲和力(DTA)预测有助于加速药物筛选并降低开发成本。为了提高DTA的预测性能和泛化能力,本文提出了一种基于离散曲率的DTA预测模型Ricci-GraphDTA,该模型将分子图和蛋白质序列建模相结合,实现了高效、准确的DTA预测。该模型由特征编码、输入表示学习和亲和力预测三部分组成。在特征编码阶段,将药物分子建模为图,引入Forman曲率来调整相邻信息聚集的权重。然后使用GIN残差网络捕获分子的局部几何和拓扑特征。利用BiLSTM对蛋白质序列进行建模,提取全局依赖特征,并通过注意机制增强捕获远程依赖和关键残基相互作用,克服了传统cnn在处理远程依赖方面的局限性。在输入表示学习阶段,将药物和蛋白质的高级表示连接起来,并通过多个非线性变换提取跨模态相互作用特征,然后将其用于亲和力预测。实验结果表明,在Davis和KIBA数据集上,Ricci-GraphDTA在各种评估指标上表现出显著的性能。进一步的冷启动实验证明了Ricci-GraphDTA在涉及未知药物或靶点的情况下具有很强的泛化能力,突出了其在现实世界药物发现应用中的潜力。平均而言,它在三个冷启动任务中实现了22.5%的MSE降低,在双冷启动设置中降低了42%以上,展示了出色的结构建模能力和鲁棒性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


