Dingyi Rong, Bozitao Zhong, Wenzhuo Zheng, Liang Hong, Ning Liu
{"title":"Autoregressive enzyme function prediction with multi-scale multi-modality fusion.","authors":"Dingyi Rong, Bozitao Zhong, Wenzhuo Zheng, Liang Hong, Ning Liu","doi":"10.1093/bib/bbaf476","DOIUrl":null,"url":null,"abstract":"<p><p>Accurate prediction of enzyme function is crucial for elucidating biological mechanisms and driving innovation across various sectors. Existing deep learning methods tend to rely solely on either sequence data or structural data and predict the Enzyme Commission (EC) number as a whole, neglecting the intrinsic hierarchical structure of EC numbers. To address these limitations, we introduce Multi-scale multi-modality Autoregressive Predictor (MAPred), a novel multi-modality and multi-scale model designed to autoregressively predict the EC number of proteins. MAPred integrates both the primary amino acid sequence and the 3D tokens of proteins, employing a dual-pathway approach to capture comprehensive protein characteristics and essential local functional sites. Additionally, MAPred utilizes an autoregressive prediction network to sequentially predict the digits of the EC number, leveraging the hierarchical organization of EC classifications. Evaluations on benchmark datasets, including New-392, Price, and New-815, demonstrate that our method outperforms existing models, marking a significant advance in the reliability and granularity of protein function prediction within bioinformatics.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"26 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12448393/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbaf476","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
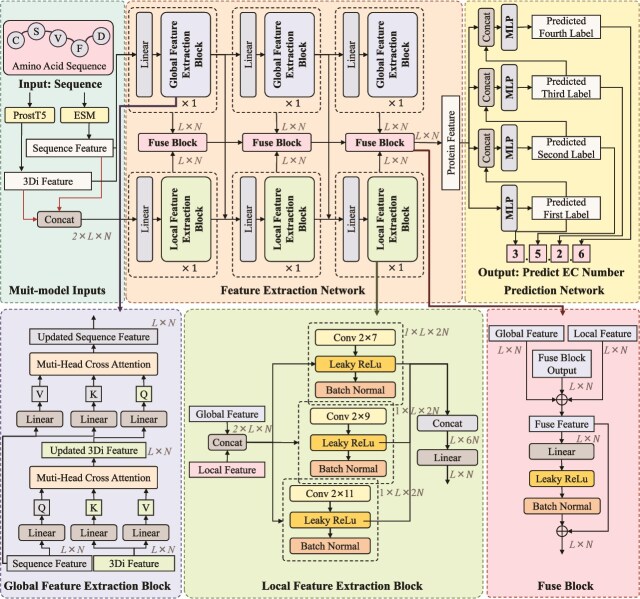
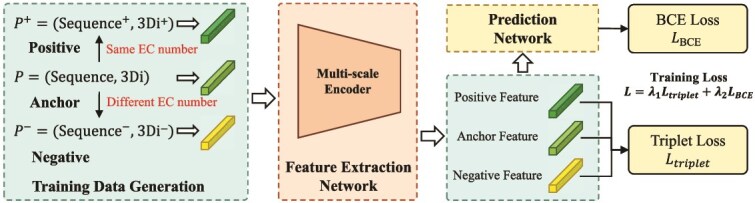
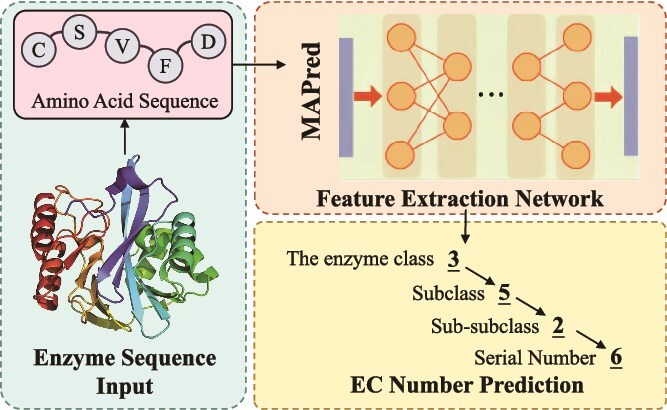
Accurate prediction of enzyme function is crucial for elucidating biological mechanisms and driving innovation across various sectors. Existing deep learning methods tend to rely solely on either sequence data or structural data and predict the Enzyme Commission (EC) number as a whole, neglecting the intrinsic hierarchical structure of EC numbers. To address these limitations, we introduce Multi-scale multi-modality Autoregressive Predictor (MAPred), a novel multi-modality and multi-scale model designed to autoregressively predict the EC number of proteins. MAPred integrates both the primary amino acid sequence and the 3D tokens of proteins, employing a dual-pathway approach to capture comprehensive protein characteristics and essential local functional sites. Additionally, MAPred utilizes an autoregressive prediction network to sequentially predict the digits of the EC number, leveraging the hierarchical organization of EC classifications. Evaluations on benchmark datasets, including New-392, Price, and New-815, demonstrate that our method outperforms existing models, marking a significant advance in the reliability and granularity of protein function prediction within bioinformatics.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


