The role of protonation and solvation in modulating the electronic structure in α- and β-D-glucosamine: A density functional theory study
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
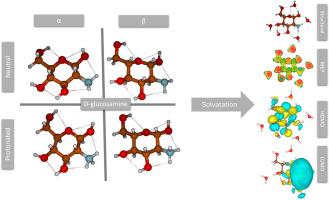
Glucosamine is a biologically relevant amino sugar that plays a key role in glycoproteins and polysaccharides such as chitin and chitosan. Despite its importance, a detailed quantum-level understanding of the electronic and conformational behavior of its α- and β-anomers under varying protonation and hydration conditions remains limited. Herein, we present a comprehensive DFT study at the B3LYP/6-31+G(d,p) level, modeling α- and β-D-glucosamine in both neutral and protonated forms with explicit hydration (1–5 water molecules). Our results show that neutral β-glucosamine is energetically more stable than its α-counterpart by 0.61 kcal/mol, consistent with its predominance in aqueous solution. Protonation induces significant electronic redistribution, with the LUMO localizing around the NH3+ group and the HOMO shifting away from it. This leads to a marked increase in the HOMO–LUMO energy gap, from 6.37 kcal/mol (neutral α-form) to a maximum of 7.32 kcal/mol (protonated β-form with 4H2O), indicating enhanced electronic stability and decreased reactivity. Explicit solvation stabilizes key hydrogen bond networks, with H-bond lengths as short as 1.51 Å in protonated systems. These findings offer new insight into how solvation and protonation shape the electronic landscape of glucosamine, with implications for its behavior in biological systems, material design, and protonation-dependent reactivity. The novelty of this study lies in combining explicit microsolvation with frontier orbital and MEP analysis across protonation states of both anomers—a previously unexplored approach.
质子化和溶剂化在调节α-和β- d -氨基葡萄糖电子结构中的作用:密度泛函理论研究。
氨基葡萄糖是一种生物相关的氨基糖,在糖蛋白和几丁质、壳聚糖等多糖中起着关键作用。尽管它很重要,但在不同质子化和水化条件下,对α-和β-异头化合物的电子和构象行为的详细量子水平理解仍然有限。在此,我们在B3LYP/6-31+G(d,p)水平上进行了全面的DFT研究,模拟了中性和质子化形式的α-和β- d -氨基葡萄糖,明确水合作用(1-5个水分子)。结果表明,中性β-氨基葡萄糖比α-氨基葡萄糖能量稳定性高0.61 kcal/mol,符合其在水溶液中的优势。质子化引起了显著的电子再分配,LUMO定位在NH3+基团周围,而HOMO则远离它。这导致HOMO-LUMO的能隙明显增加,从6.37 kcal/mol(中性α-形式)增加到7.32 kcal/mol(与4H2O质子化的β-形式),表明电子稳定性增强,反应活性降低。显式溶剂化稳定了关键的氢键网络,在质子化体系中,氢键长度短至1.51 Å。这些发现对溶剂化和质子化如何塑造葡萄糖胺的电子景观提供了新的见解,对其在生物系统中的行为、材料设计和质子依赖的反应性具有启示意义。这项研究的新颖之处在于将显式微溶剂化与前沿轨道和MEP分析结合起来,这是一种以前未探索过的方法。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


