Aminopeptidase and carboxypeptidase activity of DPP-4 on the example of peptides LPQNIPPL and LPβ3hQNIPPL
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
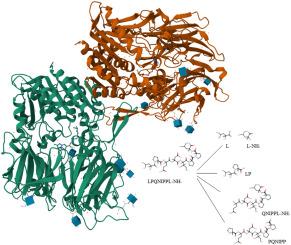
Our previous research demonstrated that LPQNIPPL and LPβ3hQNIPPL peptides are degraded in vitro by DPP-4 enzyme. Besides the expected degradation products, additional degradation products were observed on HPLC-MS chromatograms, suggesting cleavage of both the C- and N-terminal amino acids. Molecular docking provided a model for the observed carboxypeptidase, but not for aminopeptidase activity. Compared to conventional N-terminal binding at the active site, the obtained carboxypeptidase model suggested the α-amino group interaction with S2 subsite, C-terminal amino acid side chain interaction with the S1 subsite, reversed peptide backbone order and P1′ proline in a secondary peptide bond. The scissile bond position and distance relative to the active Ser630 seems to remain similar compared to conventional binding. Efficient C-terminal binding to the DPP-4 active site is reinforced by molecular dynamics results, but with altered interactions compared to molecular docking after ligand adaptation phase. Although peptide interactions and distance of scissile bond from the DPP-4 nucleophile Ser630 appears to support the cleavage model, the required nucleophilic attack angles are not confirmed. The proposed model does not allow final conclusions on nucleophilic attack stereochemistry but opens the discussion on possible DPP-4 carboxypeptidase cleavage of peptides, as well as retro/retro-inverso peptides with C-terminal Leu-NH2.
以肽LPQNIPPL和LPβ3hQNIPPL为例,分析了DPP-4的氨基肽酶和羧基肽酶活性。
我们前期的研究表明,LPQNIPPL和LPβ3hQNIPPL肽在体外被DPP-4酶降解。除了预期的降解产物外,在HPLC-MS色谱上还观察到其他降解产物,表明C端和n端氨基酸都有裂解。分子对接为观察到的羧基肽酶提供了一个模型,但没有为氨基肽酶活性提供模型。与传统的活性位点n端结合相比,所建立的羧肽酶模型表明α-氨基与S2亚位点相互作用,c -末端氨基酸侧链与S1亚位点相互作用,肽主链顺序颠倒,P1'脯氨酸在次级肽键中相互作用。与常规结合相比,相对于活性Ser630的可剪切键的位置和距离似乎保持相似。分子动力学结果增强了c端与DPP-4活性位点的有效结合,但与配体适应阶段的分子对接相比,相互作用发生了变化。虽然多肽相互作用和与DPP-4亲核试剂Ser630的可剪键距离似乎支持切割模型,但所需的亲核攻击角尚未得到证实。提出的模型不能给出亲核攻击立体化学的最终结论,但开启了对DPP-4羧肽酶可能切割肽以及具有c端Leu-NH2的逆转录/逆转录肽的讨论。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


