Whole genome sequencing of Yersinia pestis isolates from Central Asian natural plague foci revealed the role of adaptation to different hosts and environmental conditions in shaping specific genotypes.
Aigul A Abdirassilova, Duman T Yessimseit, Altynai K Kassenova, Beck Z Abdeliyev, Zauresh B Zhumadilova, Gulnara Zh Tokmurziyeva, Galina G Kovaleva, Ziyat Zh Abdel, Tatiyana V Meka-Mechenko, Saule K Umarova, Elmira Zh Begimbayeva, Sanzhar D Agzam, Vladimir L Motin, Oleg N Reva, Altyn K Rysbekova
{"title":"Whole genome sequencing of Yersinia pestis isolates from Central Asian natural plague foci revealed the role of adaptation to different hosts and environmental conditions in shaping specific genotypes.","authors":"Aigul A Abdirassilova, Duman T Yessimseit, Altynai K Kassenova, Beck Z Abdeliyev, Zauresh B Zhumadilova, Gulnara Zh Tokmurziyeva, Galina G Kovaleva, Ziyat Zh Abdel, Tatiyana V Meka-Mechenko, Saule K Umarova, Elmira Zh Begimbayeva, Sanzhar D Agzam, Vladimir L Motin, Oleg N Reva, Altyn K Rysbekova","doi":"10.1371/journal.pntd.0013533","DOIUrl":null,"url":null,"abstract":"<p><p>The genetic diversity and biovar classification of Yersinia isolates from Central Asia were investigated using whole-genome sequencing. In total, 98 isolates from natural plague foci were sequenced using the MiSeq platform. Computational pipelines were developed for accurate assembly of Y. pestis replicons, including small cryptic plasmids, and for identifying genetic polymorphisms. A panel of 99 diagnostic polymorphisms was established, enabling the distinction of dominant Medievalis isolates derived from desert and upland regions. Evidence of convergent evolution was observed in polymorphic allele distributions across genetically distinct Y. pestis biovars, Y. pseudotuberculosis, and other Y. pestis strains, likely driven by adaptation to similar environmental conditions. Genetic polymorphisms in the napA, araC, ssuA, and rhaS genes, along with transposon and CRISPR-Cas insertion patterns, were confirmed as suitable tools for identifying Y. pestis biovars, although their homoplasy suggests limited utility for phylogenetic inference. Notably, a novel cryptic plasmid, pCKF, previously associated with the strain of the population 2.MED0 from the Central-Caucasus high-altitude autonomous plague focus, was detected in a genetically distinct isolate of 2.MED1 population from the Ural-Embi region, indicating potential plasmid transfer across the 2.MED lineage. These findings emphasize the need for ongoing genomic surveillance to monitor the spread of virulence-associated genetic elements and to improve our understanding of Y. pestis evolution and ecology.</p>","PeriodicalId":49000,"journal":{"name":"PLoS Neglected Tropical Diseases","volume":"19 9","pages":"e0013533"},"PeriodicalIF":3.4000,"publicationDate":"2025-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12445494/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Neglected Tropical Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1371/journal.pntd.0013533","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"PARASITOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
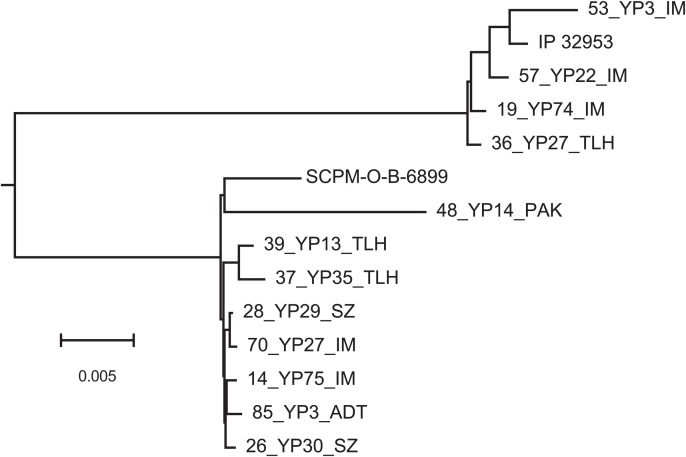
The genetic diversity and biovar classification of Yersinia isolates from Central Asia were investigated using whole-genome sequencing. In total, 98 isolates from natural plague foci were sequenced using the MiSeq platform. Computational pipelines were developed for accurate assembly of Y. pestis replicons, including small cryptic plasmids, and for identifying genetic polymorphisms. A panel of 99 diagnostic polymorphisms was established, enabling the distinction of dominant Medievalis isolates derived from desert and upland regions. Evidence of convergent evolution was observed in polymorphic allele distributions across genetically distinct Y. pestis biovars, Y. pseudotuberculosis, and other Y. pestis strains, likely driven by adaptation to similar environmental conditions. Genetic polymorphisms in the napA, araC, ssuA, and rhaS genes, along with transposon and CRISPR-Cas insertion patterns, were confirmed as suitable tools for identifying Y. pestis biovars, although their homoplasy suggests limited utility for phylogenetic inference. Notably, a novel cryptic plasmid, pCKF, previously associated with the strain of the population 2.MED0 from the Central-Caucasus high-altitude autonomous plague focus, was detected in a genetically distinct isolate of 2.MED1 population from the Ural-Embi region, indicating potential plasmid transfer across the 2.MED lineage. These findings emphasize the need for ongoing genomic surveillance to monitor the spread of virulence-associated genetic elements and to improve our understanding of Y. pestis evolution and ecology.
期刊介绍:
PLOS Neglected Tropical Diseases publishes research devoted to the pathology, epidemiology, prevention, treatment and control of the neglected tropical diseases (NTDs), as well as relevant public policy.
The NTDs are defined as a group of poverty-promoting chronic infectious diseases, which primarily occur in rural areas and poor urban areas of low-income and middle-income countries. Their impact on child health and development, pregnancy, and worker productivity, as well as their stigmatizing features limit economic stability.
All aspects of these diseases are considered, including:
Pathogenesis
Clinical features
Pharmacology and treatment
Diagnosis
Epidemiology
Vector biology
Vaccinology and prevention
Demographic, ecological and social determinants
Public health and policy aspects (including cost-effectiveness analyses).
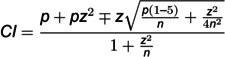

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


