Neuropeptide Y1 receptor antagonist alleviated osteoarthritis by restoring chondrocyte autophagy through PI3K/AKT/mTOR signaling pathway
IF 5.9
1区 医学
Q1 ORTHOPEDICS
引用次数: 0
Abstract
Background
Osteoarthritis (OA) is a debilitating joint disorder affecting millions worldwide, characterized by progressive cartilage degradation and chronic pain. Emerging evidence suggests that neuropeptide Y (NPY) and its Y1 receptors are involved in OA pathogenesis, although the underlying molecular mechanisms remain poorly understood. This study investigates the role of NPY/Y1R signaling in OA progression through PI3K/AKT/mTOR-mediated regulation of chondrocyte autophagy.
Methods
Human cartilage samples were collected from ten OA patients (3 male,7 female, 63–75 years old) undergoing total knee arthroplasty and graded using the Kellgren–Lawrence system. Primary chondrocytes were isolated from neonatal C57BL/6 mice and treated with NPY (0.01–5 μM) or interleukin-1β (IL-1β, 10 ng/mL) to mimic OA-like degeneration. RNA sequencing (RNA-seq) and KEGG pathway analysis were performed to identify NPY-regulated signaling pathways. In vivo, OA was induced in 8-week-old male C57BL/6 mice via destabilization of the medial meniscus (DMM) or intra-articular injections of NPY (5 μM, every 4 weeks). Mice were treated with the Y1R antagonist (0.1 μM, weekly) or vehicle control. Pain behavior was assessed using von Frey filaments and CatWalk gait analysis. Cartilage degeneration was evaluated via histology (Safranin O/Fast Green, OARSI scoring), immunofluorescence (COLII, MMP13, LC3-II, p62), and micro-CT (subchondral bone remodeling, osteophyte formation). The activation status of the PI3K/AKT/mTOR pathway and autophagy-related markers was determined via Western blotting and immunofluorescence assays under both in vitro and in vivo conditions.
Results
NPY and Y1R expression were significantly elevated in human OA cartilage compared to normal tissue. In vitro, NPY (5 μM) suppressed chondrocyte proliferation, reduced COLII expression, and increased MMP13 production. RNA-seq revealed NPY-mediated activation of the PI3K/AKT/mTOR pathway and inhibition of autophagy-related genes. NPY treatment enhanced the phosphorylation levels of PI3K, AKT, and mTOR, while concurrently decreasing LC3II expression and increasing p62 accumulation. The Y1R antagonist reversed these effects, restoring autophagy and attenuating cartilage degradation. In vivo, NPY injections induced OA-like changes, including cartilage thinning, osteophyte formation, and mechanical allodynia. Y1R antagonist treatment mitigated these effects, improving gait parameters and reducing subchondral bone sclerosis. Immunofluorescence confirmed that Y1R inhibition decreased PI3K/AKT/mTOR signaling and enhanced autophagy in chondrocytes.
Conclusion
This study demonstrates that NPY/Y1R signaling exacerbates OA progression through PI3K/AKT/mTOR-mediated suppression of chondrocyte autophagy. Pharmacological inhibition of Y1R emerges as a novel therapeutic strategy, effectively targeting both cartilage degeneration and pain, with potential disease-modifying effects on osteoarthritis progression.
The translational potential of this article
This study highlights the NPY Y1 receptor as a promising therapeutic target for OA by demonstrating its role in modulating chondrocyte autophagy via the PI3K/AKT/mTOR pathway. The results support the development of Y1R antagonists as novel OA therapeutics. This work bridges molecular discovery to potential clinical application, offering hope for a transformative approach to OA management.
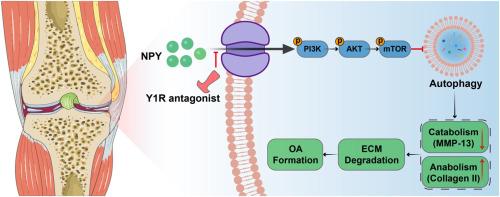
神经肽Y1受体拮抗剂通过PI3K/AKT/mTOR信号通路恢复软骨细胞自噬,减轻骨关节炎
骨关节炎(OA)是一种影响全球数百万人的衰弱性关节疾病,其特征是进行性软骨退化和慢性疼痛。新的证据表明神经肽Y (NPY)及其Y1受体参与OA发病机制,尽管其潜在的分子机制尚不清楚。本研究通过PI3K/AKT/ mtor介导的软骨细胞自噬调节,探讨NPY/Y1R信号在OA进展中的作用。方法对10例OA患者(男3例,女7例,年龄63 ~ 75岁)进行全膝关节置换术,并采用Kellgren-Lawrence评分系统进行评分。从新生C57BL/6小鼠中分离原代软骨细胞,用NPY (0.01 ~ 5 μM)或白细胞介素-1β (IL-1β, 10 ng/mL)处理,模拟a样变性。通过RNA测序(RNA-seq)和KEGG通路分析鉴定npy调控的信号通路。在体内,通过破坏内侧半月板(DMM)或关节内注射NPY (5 μM,每4周),在8周龄雄性C57BL/6小鼠中诱导OA。小鼠用Y1R拮抗剂(0.1 μM,每周)或对照治疗。采用von Frey纤维和CatWalk步态分析评估疼痛行为。通过组织学(Safranin O/Fast Green, OARSI评分),免疫荧光(COLII, MMP13, LC3-II, p62)和显微ct(软骨下骨重塑,骨赘形成)评估软骨退行性变。在体外和体内条件下,通过Western blotting和免疫荧光法检测PI3K/AKT/mTOR通路和自噬相关标志物的激活状态。结果骨性关节炎软骨中snpy和Y1R的表达明显高于正常组织。在体外,NPY (5 μM)抑制软骨细胞增殖,降低COLII表达,增加MMP13的产生。RNA-seq显示npy介导的PI3K/AKT/mTOR通路的激活和自噬相关基因的抑制。NPY处理提高了PI3K、AKT和mTOR的磷酸化水平,同时降低了LC3II的表达,增加了p62的积累。Y1R拮抗剂逆转了这些作用,恢复自噬并减轻软骨降解。在体内,NPY注射诱导oa样改变,包括软骨变薄、骨赘形成和机械异常性痛。Y1R拮抗剂治疗减轻了这些影响,改善了步态参数并减少了软骨下骨硬化。免疫荧光证实,Y1R抑制降低了PI3K/AKT/mTOR信号传导,增强了软骨细胞的自噬。结论本研究表明NPY/Y1R信号通过PI3K/AKT/ mtor介导的软骨细胞自噬抑制加速OA进展。Y1R的药理抑制是一种新的治疗策略,有效地针对软骨变性和疼痛,对骨关节炎的进展具有潜在的疾病改善作用。本研究通过证明NPY Y1受体通过PI3K/AKT/mTOR通路调节软骨细胞自噬的作用,强调了NPY Y1受体作为OA的一个有希望的治疗靶点。该结果支持开发Y1R拮抗剂作为新型OA治疗药物。这项工作将分子发现与潜在的临床应用联系起来,为OA管理的变革方法提供了希望。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Orthopaedic Translation
Medicine-Orthopedics and Sports Medicine
CiteScore
11.80
自引率
13.60%
发文量
91
审稿时长
29 days
期刊介绍:
The Journal of Orthopaedic Translation (JOT) is the official peer-reviewed, open access journal of the Chinese Speaking Orthopaedic Society (CSOS) and the International Chinese Musculoskeletal Research Society (ICMRS). It is published quarterly, in January, April, July and October, by Elsevier.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


