Thermodynamic and structural insights into full-length amyloid-β42 dimerization using enhanced sampling and MD-based techniques
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
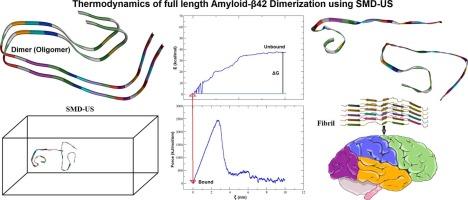
Amyloid-β (Aβ) peptide aggregation into oligomers is a key pathological feature of Alzheimer's disease (AD). Of early aggregates, the Aβ dimer is the most basic neurotoxic species. Despite numerous computational studies that offer helpful information about the structural and energetic properties of Aβ dimers, comprehensive studies that integrate binding energetics with conformational dynamics of the full-length Aβ42 dimer under near-physiological conditions remain scarce. In the present work, we used molecular docking, molecular dynamics (MD), steered MD (SMD), and umbrella sampling (US) approaches to examine the different stages of full-length Aβ42 dimerization. The free energy of dimerization, predicted as −37.57 kcal/mol by the SMD-US method from crystallographic aggregate (PDB ID: 5OQV), reflects thermodynamically favorable binding of monomer peptides. We also identified certain particular non-covalent interactions that stabilize the dimer structure and oligomerization process. These findings offer mechanistic insights into Aβ42 oligomerization and deepen our understanding of amyloid aggregation in AD.
热力学和结构的见解全长淀粉样蛋白-β42二聚化使用增强采样和md为基础的技术
淀粉样蛋白-β (a β)肽聚集成低聚物是阿尔茨海默病(AD)的一个关键病理特征。在早期聚集体中,Aβ二聚体是最基本的神经毒性物质。尽管大量的计算研究提供了有关Aβ二聚体的结构和能量特性的有用信息,但在近生理条件下将全长Aβ42二聚体的结合能学与构象动力学相结合的综合研究仍然很少。在本研究中,我们使用分子对接、分子动力学(MD)、定向MD (SMD)和伞式采样(US)方法来研究全长Aβ42二聚化的不同阶段。晶体聚合体(PDB ID: 5OQV)的SMD-US法预测二聚化的自由能为−37.57 kcal/mol,反映了单体肽在热力学上的良好结合。我们还确定了某些特定的非共价相互作用,稳定二聚体结构和寡聚化过程。这些发现提供了a - β42寡聚化的机制见解,并加深了我们对AD中淀粉样蛋白聚集的理解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


