{"title":"Ligand B-Factor Index: A Metric for Prioritizing Protein-Ligand Complexes in Docking.","authors":"Liliana Halip, Cristian Neanu, Sorin Avram","doi":"10.1002/minf.70010","DOIUrl":null,"url":null,"abstract":"<p><p>Docking is a structure-based cheminformatics tool broadly employed in early drug discovery. Based on the tridimensional structure of the protein target, docking is used to predict the binding interactions between the protein and a ligand, estimate the corresponding binding affinity, or perform virtual screenings (VSs) to identify new active compounds. This study introduces the ligand B-factor index (LBI), a novel computational metric for prioritizing protein-ligand complexes for docking. Unlike other metrics, LBI directly compares atomic displacements in the ligand and binding site. LBI is defined as the ratio of the median atomic B-factor of the binding site to that of the bound ligand. Using the comparative assessment of scoring functions (CASF-2016) dataset, we evaluated the effectiveness of LBI in guiding the selection of protein-ligand complexes to enhance docking performance. Our results show a moderate correlation (Spearman ρ ~ 0.48) between LBI and the experimental binding affinities, outperforming several docking scoring functions. Additionally, LBI correlates with improved redocking success (root mean square deviation < 2 Å), underlying the significance of a ligand-focused metric. While LBI outperforms other metrics such as the protein B-factor index and resolution, its utility in VS docking remains to be further investigated. LBI is easy to compute, interpretable, applicable in structure-based cheminformatics, and freely available for calculation at https://chembioinf.ro/tool-bi-computing.html.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"44 9","pages":"e202500127"},"PeriodicalIF":3.1000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12423484/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.70010","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
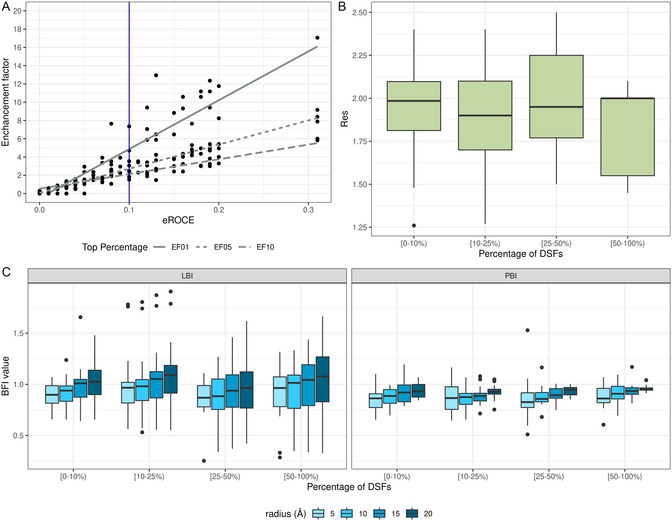
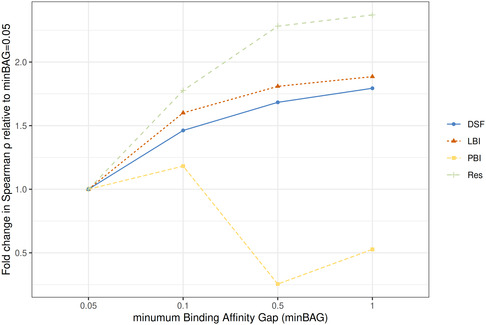
Docking is a structure-based cheminformatics tool broadly employed in early drug discovery. Based on the tridimensional structure of the protein target, docking is used to predict the binding interactions between the protein and a ligand, estimate the corresponding binding affinity, or perform virtual screenings (VSs) to identify new active compounds. This study introduces the ligand B-factor index (LBI), a novel computational metric for prioritizing protein-ligand complexes for docking. Unlike other metrics, LBI directly compares atomic displacements in the ligand and binding site. LBI is defined as the ratio of the median atomic B-factor of the binding site to that of the bound ligand. Using the comparative assessment of scoring functions (CASF-2016) dataset, we evaluated the effectiveness of LBI in guiding the selection of protein-ligand complexes to enhance docking performance. Our results show a moderate correlation (Spearman ρ ~ 0.48) between LBI and the experimental binding affinities, outperforming several docking scoring functions. Additionally, LBI correlates with improved redocking success (root mean square deviation < 2 Å), underlying the significance of a ligand-focused metric. While LBI outperforms other metrics such as the protein B-factor index and resolution, its utility in VS docking remains to be further investigated. LBI is easy to compute, interpretable, applicable in structure-based cheminformatics, and freely available for calculation at https://chembioinf.ro/tool-bi-computing.html.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


