Neonatal liver-derived FTH1-enriched extracellular vesicles attenuate ferroptosis and ameliorate MASLD pathogenesis
IF 8.2
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD), a leading cause of chronic liver pathology, lacks effective therapies. This study identifies ferroptosis—a lipid peroxidation-driven, iron-dependent form of cell death—as a central pathogenic mechanism in MASLD. Integrative proteomic and histopathological analyses of human and murine MASLD livers revealed marked ferroptosis activation, characterized by dysregulated iron metabolism (reduced FTH1 and GPX4; elevated ACSL4) and oxidative stress. To address this, neonatal liver-derived extracellular vesicles (EVnew) enriched with FTH1, were evaluated as a therapeutic strategy. EVnew exhibited superior biodistribution to adult-derived EVs (EVadult), with liver-specific tropism and proteomic enrichment of FTH1, mitochondrial proteins, and glutathione regulators. In MASLD models, EVnew administration attenuated hepatic injury, steatosis, and fibrosis, outperforming EVadult by restoring iron homeostasis via FTH1-mediated suppression of lipid peroxidation. Specifically, EVnew upregulated NRF2 and GPX4 while downregulating ACSL4 and HSP60. Neutralizing FTH1 abolished the protective effects of EVnew in vitro, confirming that FTH1 is the key mediator. These findings establish ferroptosis as a therapeutic target in MASLD and highlight the dual capacity of EVnew to inhibit ferroptosis. This work pioneers xenogeneic EVs-based therapy for metabolic liver diseases, offering a novel multifaceted intervention with translational potential.
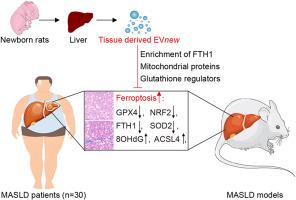
新生儿肝脏来源的富fth1细胞外囊泡减轻铁下垂和改善MASLD发病机制。
代谢功能障碍相关脂肪变性肝病(MASLD)是慢性肝脏病理的主要原因,缺乏有效的治疗方法。本研究确定了铁中毒——一种脂质过氧化驱动的、铁依赖的细胞死亡形式——是MASLD的主要致病机制。对人和小鼠MASLD肝脏的综合蛋白质组学和组织病理学分析显示,明显的铁凋亡激活,其特征是铁代谢失调(FTH1和GPX4减少,ACSL4升高)和氧化应激。为了解决这个问题,新生儿肝源性细胞外囊泡(EVnew)富含FTH1,作为一种治疗策略进行了评估。EVnew在成人源EVs (EVadult)中表现出优越的生物分布,具有肝脏特异性趋向性和FTH1、线粒体蛋白和谷胱甘肽调节因子的蛋白质组学富集。在MASLD模型中,EVnew通过fth1介导的脂质过氧化抑制恢复铁稳态,减轻了肝损伤、脂肪变性和纤维化,优于EVadult。具体来说,EVnew上调NRF2和GPX4,下调ACSL4和HSP60。在体外,中和FTH1可消除EVnew的保护作用,证实FTH1是关键的介质。这些发现确立了铁下垂作为MASLD的治疗靶点,并强调了EVnew抑制铁下垂的双重能力。这项工作开创了代谢性肝病的异种evs治疗,提供了一种具有转化潜力的新的多方面干预。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Free Radical Biology and Medicine
医学-内分泌学与代谢
CiteScore
14.00
自引率
4.10%
发文量
850
审稿时长
22 days
期刊介绍:
Free Radical Biology and Medicine is a leading journal in the field of redox biology, which is the study of the role of reactive oxygen species (ROS) and other oxidizing agents in biological systems. The journal serves as a premier forum for publishing innovative and groundbreaking research that explores the redox biology of health and disease, covering a wide range of topics and disciplines. Free Radical Biology and Medicine also commissions Special Issues that highlight recent advances in both basic and clinical research, with a particular emphasis on the mechanisms underlying altered metabolism and redox signaling. These Special Issues aim to provide a focused platform for the latest research in the field, fostering collaboration and knowledge exchange among researchers and clinicians.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


