Ke Zeng, Yuqi Zhu, Zhongxin Han, Siyi Xiong, Yan Zhao, Zilong Xiao, Yingchao Xie, Shiyu Jin, Tingru Dong, Lan Lan, Weiwei Liu, Yongzhong Du, Cuiping Guan, Xiao Yu, Xiuzu Song
{"title":"NLRP3 autophagic degradation disruption in melanocytes contributes to vitiligo development","authors":"Ke Zeng, Yuqi Zhu, Zhongxin Han, Siyi Xiong, Yan Zhao, Zilong Xiao, Yingchao Xie, Shiyu Jin, Tingru Dong, Lan Lan, Weiwei Liu, Yongzhong Du, Cuiping Guan, Xiao Yu, Xiuzu Song","doi":"10.1038/s41418-025-01578-5","DOIUrl":null,"url":null,"abstract":"<p>NLRP3 functions as a critical intracellular danger sensor for inflammasome activation, playing a crucial role in autoimmune diseases. Vitiligo progression has been linked to NLRP3, yet its specific involvement in melanocytes of vitiligo remains poorly understood. In this study, we demonstrate that NLRP3 expression is significantly upregulated in the melanocytes of vitiligo patients and melanoma-Treg-induced vitiligo mouse model. Genetic knockout of NLRP3 effectively alleviates vitiligo progression in these mice. Our mechanistic investigations reveal that the downregulation of the E3 ligase β-TrCP1 in vitiligo melanocytes decreases K27-linked ubiquitination levels of NLRP3, which in turn weakens its interaction with the autophagy receptor NDP52. This disruption impairs the selective autophagic degradation of NLRP3, leading to hyperactivation of inflammation and pyroptosis in melanocytes, thereby accelerating vitiligo pathogenesis. Notably, melanocyte-specific knockdown of NLRP3 using lysine-proline-valine (KPV)-modified deformable liposomes (KPV-Lipos) carrying <i>Nlrp3</i> shRNA significantly alleviates vitiligo development. This study elucidates the mechanism by which autophagy dysfunction mediated excessive NLRP3 inflammasome activation in melanocytes contributes to vitiligo pathogenesis, highlighting potential therapeutic strategies targeting these pathways for the treatment of vitiligo and other pigment-related skin diseases.</p><figure><p><b>Overview of disrupted NLRP3 autophagic degradation in vitiligo melanocytes</b>. In healthy melanocytes, NLRP3 expression is upregulated when subjected to oxidative stress, along with an increase in the E3 ligase β-TrCP1, which enhances the K27-linked ubiquitination of NLRP3 and further strengthens its binding to the autophagy receptor protein NDP52, thus effectively suppressing the excessive inflammatory response. Whereas in the melanocytes of vitiligo patients, decreased expression of β-TrCP1 leads to downregulation of K27-linked ubiquitination in NLRP3, thus inhibiting its autophagic degradation. The persistent activation of NLRP3 in vitiligo melanocytes promotes the cleavage of pro-IL-1β and GSDMD. GSDMD-N subsequently forms pores on the cell membrane, which causes the release of IL-1β and results in melanocyte pyroptosis. In our study, we utilize KPV-Lipos with <i>Nlrp3</i> shRNA to precisely knockdown NLRP3 expression in melanocytes and effectively alleviate vitiligo development, which provide a potentially promising strategy for the treatment of vitiligo. MC, melanocytes.</p></figure>","PeriodicalId":9731,"journal":{"name":"Cell Death and Differentiation","volume":"74 1","pages":""},"PeriodicalIF":15.4000,"publicationDate":"2025-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death and Differentiation","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41418-025-01578-5","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
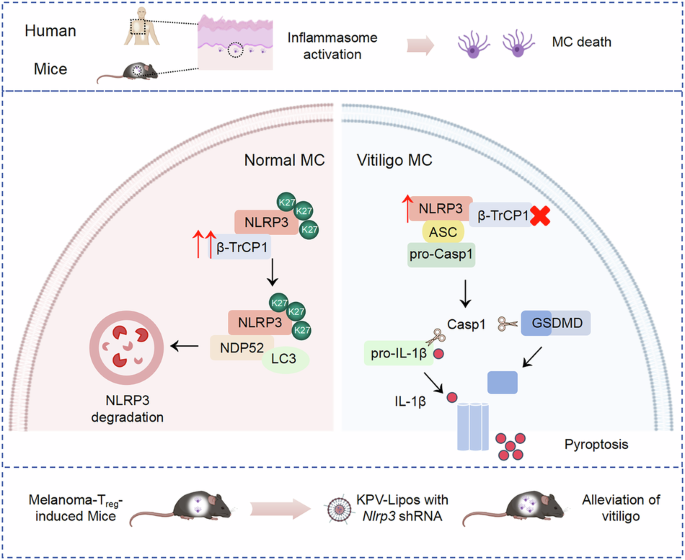
NLRP3 functions as a critical intracellular danger sensor for inflammasome activation, playing a crucial role in autoimmune diseases. Vitiligo progression has been linked to NLRP3, yet its specific involvement in melanocytes of vitiligo remains poorly understood. In this study, we demonstrate that NLRP3 expression is significantly upregulated in the melanocytes of vitiligo patients and melanoma-Treg-induced vitiligo mouse model. Genetic knockout of NLRP3 effectively alleviates vitiligo progression in these mice. Our mechanistic investigations reveal that the downregulation of the E3 ligase β-TrCP1 in vitiligo melanocytes decreases K27-linked ubiquitination levels of NLRP3, which in turn weakens its interaction with the autophagy receptor NDP52. This disruption impairs the selective autophagic degradation of NLRP3, leading to hyperactivation of inflammation and pyroptosis in melanocytes, thereby accelerating vitiligo pathogenesis. Notably, melanocyte-specific knockdown of NLRP3 using lysine-proline-valine (KPV)-modified deformable liposomes (KPV-Lipos) carrying Nlrp3 shRNA significantly alleviates vitiligo development. This study elucidates the mechanism by which autophagy dysfunction mediated excessive NLRP3 inflammasome activation in melanocytes contributes to vitiligo pathogenesis, highlighting potential therapeutic strategies targeting these pathways for the treatment of vitiligo and other pigment-related skin diseases.
Overview of disrupted NLRP3 autophagic degradation in vitiligo melanocytes. In healthy melanocytes, NLRP3 expression is upregulated when subjected to oxidative stress, along with an increase in the E3 ligase β-TrCP1, which enhances the K27-linked ubiquitination of NLRP3 and further strengthens its binding to the autophagy receptor protein NDP52, thus effectively suppressing the excessive inflammatory response. Whereas in the melanocytes of vitiligo patients, decreased expression of β-TrCP1 leads to downregulation of K27-linked ubiquitination in NLRP3, thus inhibiting its autophagic degradation. The persistent activation of NLRP3 in vitiligo melanocytes promotes the cleavage of pro-IL-1β and GSDMD. GSDMD-N subsequently forms pores on the cell membrane, which causes the release of IL-1β and results in melanocyte pyroptosis. In our study, we utilize KPV-Lipos with Nlrp3 shRNA to precisely knockdown NLRP3 expression in melanocytes and effectively alleviate vitiligo development, which provide a potentially promising strategy for the treatment of vitiligo. MC, melanocytes.
期刊介绍:
Mission, vision and values of Cell Death & Differentiation:
To devote itself to scientific excellence in the field of cell biology, molecular biology, and biochemistry of cell death and disease.
To provide a unified forum for scientists and clinical researchers
It is committed to the rapid publication of high quality original papers relating to these subjects, together with topical, usually solicited, reviews, meeting reports, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


