{"title":"Large-scale composite hypothesis testing procedure for omics data analyses.","authors":"Annaïg De Walsche, Franck Gauthier, Nathalie Boissot, Alain Charcosset, Tristan Mary-Huard","doi":"10.1093/nargab/lqaf118","DOIUrl":null,"url":null,"abstract":"<p><p>Composite hypothesis testing using summary statistics is a well-established approach for assessing the effect of a single marker or gene across multiple traits or omics levels. Numerous procedures have been developed for this task and have been successfully applied to identify complex patterns of association between traits, conditions, or phenotypes. However, existing methods often struggle with scalability in large datasets or fail to account for dependencies between traits or omics levels, limiting their ability to control false positives effectively. To overcome these challenges, we present the qch_copula approach, which integrates mixture models with a copula function to capture dependencies between traits or omics and provides rigorously defined <i>P</i>-values for any composite hypothesis. Through a comprehensive benchmark against eight state-of-the-art methods, we demonstrate that qch_copula controls Type I error rates effectively while enhancing the detection of joint association patterns. Compared to other mixture model-based approaches, our method notably reduces memory usage during the EM algorithm, allowing the analysis of up to 20 traits and 10<sup>5</sup>-10<sup>6</sup> markers. The effectiveness of qch_copula is further validated through two application cases in human and plant genetics. The method is available in the R package qch, accessible on CRAN.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"7 3","pages":"lqaf118"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12412788/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqaf118","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
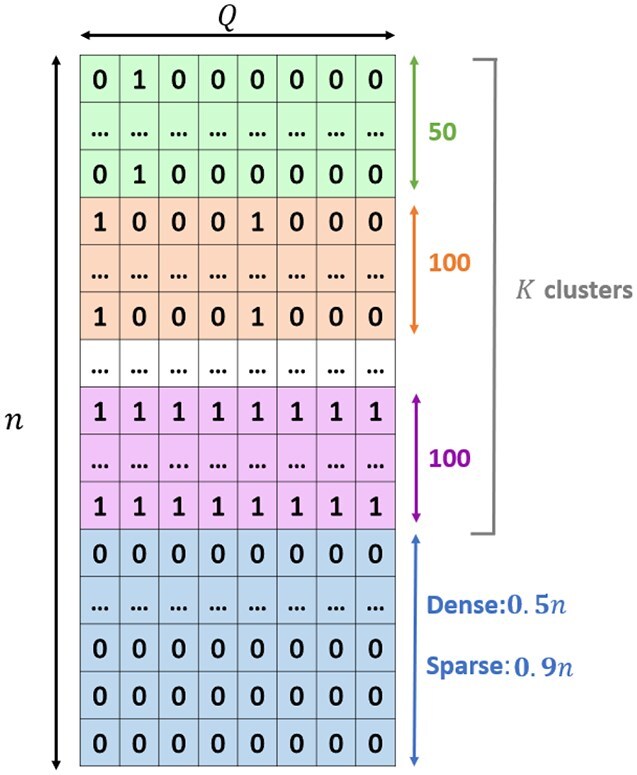
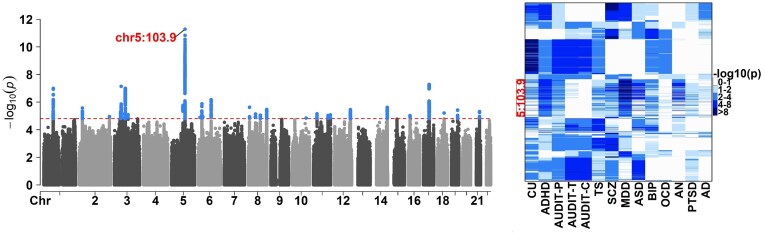
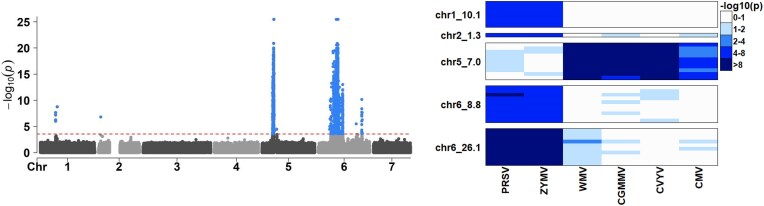
Composite hypothesis testing using summary statistics is a well-established approach for assessing the effect of a single marker or gene across multiple traits or omics levels. Numerous procedures have been developed for this task and have been successfully applied to identify complex patterns of association between traits, conditions, or phenotypes. However, existing methods often struggle with scalability in large datasets or fail to account for dependencies between traits or omics levels, limiting their ability to control false positives effectively. To overcome these challenges, we present the qch_copula approach, which integrates mixture models with a copula function to capture dependencies between traits or omics and provides rigorously defined P-values for any composite hypothesis. Through a comprehensive benchmark against eight state-of-the-art methods, we demonstrate that qch_copula controls Type I error rates effectively while enhancing the detection of joint association patterns. Compared to other mixture model-based approaches, our method notably reduces memory usage during the EM algorithm, allowing the analysis of up to 20 traits and 105-106 markers. The effectiveness of qch_copula is further validated through two application cases in human and plant genetics. The method is available in the R package qch, accessible on CRAN.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


