{"title":"GCNMF-SDA: predicting snoRNA-disease associations based on graph convolution and non-negative matrix factorization.","authors":"Yaowu Zhang, Xiu Jin, Xiaodan Zhang","doi":"10.1093/bib/bbaf453","DOIUrl":null,"url":null,"abstract":"<p><p>Small nucleolar RNAs (snoRNAs) play crucial roles in a wide range of biological processes, and studying their association with diseases can enhance our understanding of disease pathogenesis. Nevertheless, current knowledge of these associations is limited traditional biological experiments are both costly and time-consuming. Consequently, developing efficient computational methods is essential for predicting potential snoRNA-disease associations. We propose a novel prediction method based on non-negative matrix factorization and graph convolution for predicting snoRNA-disease associations (GCNMF-SDA). First, five different types of similarity information from snoRNA and disease entities are introduced to fully mine and refine the feature information. Then the snoRNA and disease similarity networks are integrated using nonlinearity approach Similarity Network Fusion (SNF), while the weighted K nearest known neighbors (WKNKN) algorithm is applied to optimize the snoRNA-disease association matrix. Following this, the graph convolution module and the non-negative matrix factorization module extract disease features and snoRNA features, respectively. After extracting these features, they are combined into a composite feature vector for each snoRNA-disease pair. Finally, the composite feature vectors along with their corresponding labels, are input into a multilayer perceptron for training. Our experiments, conducted using a rigorous five-fold cross-validation approach, reveal that the GCNMF-SDA model achieves an impressive area under the receiver operating characteristic curve (AUC-ROC) of 0.9659 and an area under the precision-recall curve (AUC-PR) of 0.9522. Furthermore, most of the novel associations identified by GCNMF-SDA were validated through case studies, underscoring the method's reliability in predicting potential relationships between snoRNAs and diseases.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"26 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409419/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbaf453","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
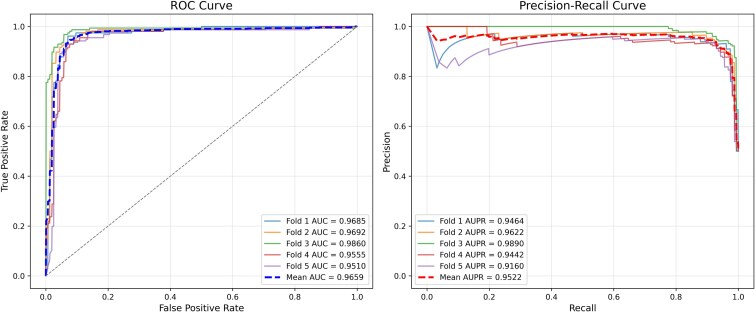
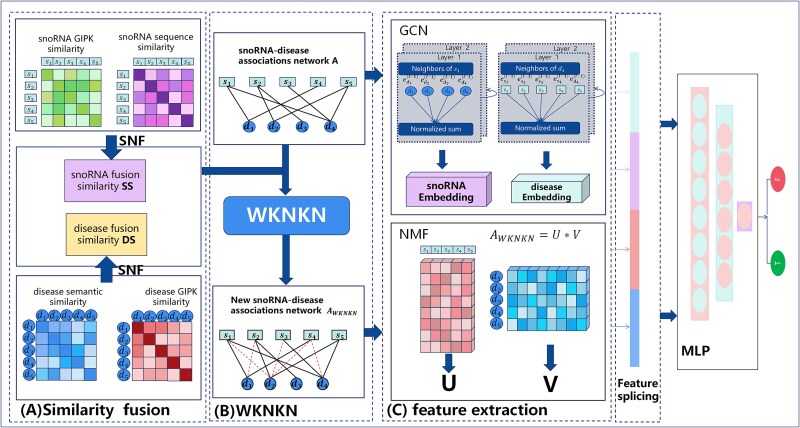
Small nucleolar RNAs (snoRNAs) play crucial roles in a wide range of biological processes, and studying their association with diseases can enhance our understanding of disease pathogenesis. Nevertheless, current knowledge of these associations is limited traditional biological experiments are both costly and time-consuming. Consequently, developing efficient computational methods is essential for predicting potential snoRNA-disease associations. We propose a novel prediction method based on non-negative matrix factorization and graph convolution for predicting snoRNA-disease associations (GCNMF-SDA). First, five different types of similarity information from snoRNA and disease entities are introduced to fully mine and refine the feature information. Then the snoRNA and disease similarity networks are integrated using nonlinearity approach Similarity Network Fusion (SNF), while the weighted K nearest known neighbors (WKNKN) algorithm is applied to optimize the snoRNA-disease association matrix. Following this, the graph convolution module and the non-negative matrix factorization module extract disease features and snoRNA features, respectively. After extracting these features, they are combined into a composite feature vector for each snoRNA-disease pair. Finally, the composite feature vectors along with their corresponding labels, are input into a multilayer perceptron for training. Our experiments, conducted using a rigorous five-fold cross-validation approach, reveal that the GCNMF-SDA model achieves an impressive area under the receiver operating characteristic curve (AUC-ROC) of 0.9659 and an area under the precision-recall curve (AUC-PR) of 0.9522. Furthermore, most of the novel associations identified by GCNMF-SDA were validated through case studies, underscoring the method's reliability in predicting potential relationships between snoRNAs and diseases.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


