Theoretical investigation on enhancing quantum efficiency of TADF molecules through modulating substituent effects
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Thermally activated delayed fluorescence (TADF) molecules with a Donor-Acceptor-Donor (D-A-D) architecture have attracted extensive attention by virtue of their widespread applications in organic light-emitting diodes (OLEDs). In this work, The time-dependent density functional theory (TD-DFT), combined with the path integral approach to dynamics considering the Herzberg-Teller effect, was employed to theoretically calculate the decay rates of the designed five molecules. Molecule II with meta-substituted has a high internal conversion rate, resulting in a significantly lower delayed fluorescence quantum efficiency compared to molecule I with para-substitute. Additionally, the introduction of weak electron-donating substituents on the acceptor unit effectively reduce the energy gap of the excited states and increase the spin-orbit coupling (SOC) constant, thereby enhancing the rates of intersystem crossing (ISC) and reverse intersystem crossing (RISC), which significantly improves the delayed fluorescence quantum efficiency. This research will contribute to the design of novel and highly efficient TADF materials.
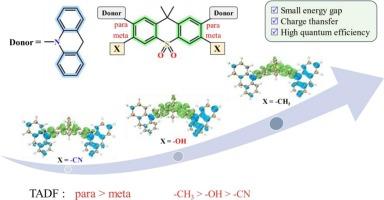
通过调节取代基效应提高TADF分子量子效率的理论研究
具有供体-受体-供体(D-A-D)结构的热激活延迟荧光(TADF)分子由于其在有机发光二极管(oled)中的广泛应用而引起了广泛的关注。本文采用时间依赖的密度泛函理论(TD-DFT),结合考虑Herzberg-Teller效应的动力学路径积分方法,从理论上计算了所设计的五种分子的衰变率。具有元取代的分子II具有较高的内部转化率,导致延迟荧光量子效率明显低于具有副取代的分子I。此外,在受体单元上引入弱给电子取代基,有效地减小了激发态的能隙,增加了自旋轨道耦合(SOC)常数,从而提高了系统间交叉(ISC)和反向系统间交叉(RISC)的速率,显著提高了延迟荧光量子效率。本研究将有助于设计新型高效的TADF材料。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


