Qicheng Bei, Nathan L R Williams, Laura E Furtado, Daria Di Blasi, Jelani Williams, Vanda Brotas, Glen Tarran, Andrew P Rees, Chris Bowler, Jed A Fuhrman
{"title":"Quantitative metagenomics for marine prokaryotes and photosynthetic eukaryotes.","authors":"Qicheng Bei, Nathan L R Williams, Laura E Furtado, Daria Di Blasi, Jelani Williams, Vanda Brotas, Glen Tarran, Andrew P Rees, Chris Bowler, Jed A Fuhrman","doi":"10.1093/ismeco/ycaf131","DOIUrl":null,"url":null,"abstract":"<p><p>High-throughput sequencing has provided unprecedented insights into microbial biodiversity in marine and other ecosystems. However, most sequencing-based studies report only relative (compositional) rather than absolute abundance, limiting their application in ecological modeling and biogeochemical analyses. Here, we present a metagenomic protocol incorporating genomic internal standards to quantify the absolute abundances of prokaryotes and eukaryotic phytoplankton, which together form the base of the marine food web, in unfractionated seawater. We applied this method to surface waters collected across 50°N to 40°S during the 29<sup>th</sup> Atlantic Meridional Transect. Using the single-copy <i>recA</i> gene, we estimated an average bacterial abundance of 1.0 × 10<sup>9</sup> haploid genome equivalents per liter. Leveraging a recent report that the <i>psbO</i> gene is typically single-copy in phytoplankton, we also quantified eukaryotic phytoplankton. Metagenomic estimates closely aligned with flow cytometry data for cyanobacteria (slope = 1.03, Pearson's <i>r</i> = 0.89) and eukaryotic phytoplankton (slope = 0.72, Pearson's <i>r</i> = 0.84). Compared to flow cytometry, taxonomic resolution for nano- and picoeukaryotes was greatly improved. Estimates for diatoms, dinoflagellates, and <i>Trichodesmium</i> were considerably higher than microscopy counts, likely reflecting microscopy undercounts and potential ploidy variation. These findings highlight the value of absolute quantification by metagenomics and offer a robust framework for quantitative assessments in microbial oceanography.</p>","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":"5 1","pages":"ycaf131"},"PeriodicalIF":6.1000,"publicationDate":"2025-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12378644/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/ismeco/ycaf131","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
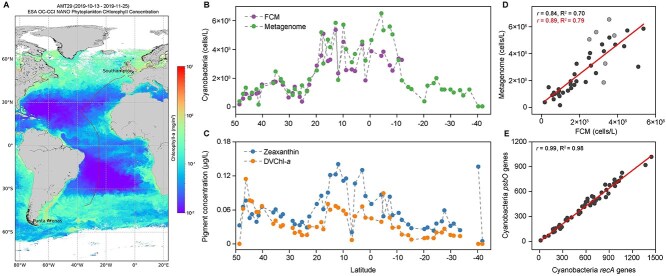
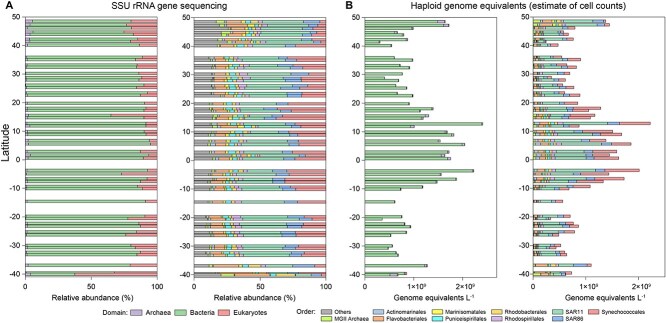
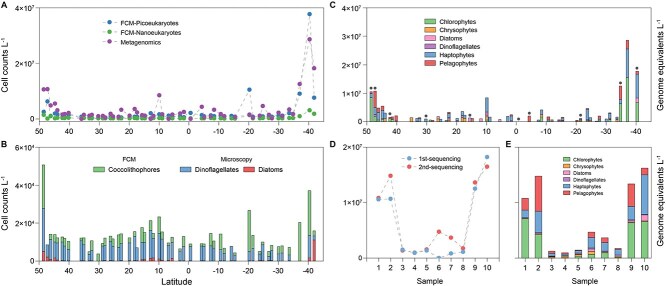
High-throughput sequencing has provided unprecedented insights into microbial biodiversity in marine and other ecosystems. However, most sequencing-based studies report only relative (compositional) rather than absolute abundance, limiting their application in ecological modeling and biogeochemical analyses. Here, we present a metagenomic protocol incorporating genomic internal standards to quantify the absolute abundances of prokaryotes and eukaryotic phytoplankton, which together form the base of the marine food web, in unfractionated seawater. We applied this method to surface waters collected across 50°N to 40°S during the 29th Atlantic Meridional Transect. Using the single-copy recA gene, we estimated an average bacterial abundance of 1.0 × 109 haploid genome equivalents per liter. Leveraging a recent report that the psbO gene is typically single-copy in phytoplankton, we also quantified eukaryotic phytoplankton. Metagenomic estimates closely aligned with flow cytometry data for cyanobacteria (slope = 1.03, Pearson's r = 0.89) and eukaryotic phytoplankton (slope = 0.72, Pearson's r = 0.84). Compared to flow cytometry, taxonomic resolution for nano- and picoeukaryotes was greatly improved. Estimates for diatoms, dinoflagellates, and Trichodesmium were considerably higher than microscopy counts, likely reflecting microscopy undercounts and potential ploidy variation. These findings highlight the value of absolute quantification by metagenomics and offer a robust framework for quantitative assessments in microbial oceanography.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


