Laura van Weesep, Rıza Özçelik, Marloes Pennings, Emanuele Criscuolo, Christian Ottmann, Luc Brunsveld and Francesca Grisoni
{"title":"Identifying 14-3-3 interactome binding sites with deep learning","authors":"Laura van Weesep, Rıza Özçelik, Marloes Pennings, Emanuele Criscuolo, Christian Ottmann, Luc Brunsveld and Francesca Grisoni","doi":"10.1039/D5DD00132C","DOIUrl":null,"url":null,"abstract":"<p >Protein–protein interactions are at the heart of biological processes. Understanding how proteins interact is key for deciphering their roles in health and disease, and for therapeutic interventions. However, identifying protein interaction sites, especially for intrinsically disordered proteins, is challenging. Here, we developed a deep learning framework to predict potential protein binding sites to 14-3-3 – a ‘central hub’ protein holding a key role in cellular signaling networks. After systematically testing multiple deep learning approaches to predict sequence binding to 14-3-3, we developed an ensemble model that achieved a 75% balanced accuracy on external sequences. Our approach was applied prospectively to identify putative binding sites across medically relevant proteins (ranging from highly structured to intrinsically disordered) for a total of approximately 300 sequences. The top eight predicted peptide sequences were experimentally validated in the wet-lab, and binding to 14-3-3 was confirmed for five out of eight sequences (<em>K</em><small><sub>d</sub></small> ranging from 1.6 ± 0.1 μM to 70 ± 5 μM). The relevance of our results was further confirmed by X-ray crystallography and molecular dynamics simulations. These sequences represent potential new binding sites within the 14-3-3 interactome (<em>e.g.</em>, relating to Alzheimer's disease as the binding to tau is not the new part), and provide opportunities to investigate their functional relevance. Our results highlight the ability of deep learning to capture intricate patterns underlying protein–protein interactions, even for challenging cases like intrinsically disordered proteins. To further the understanding and targeting of 14-3-3/protein interactions, our model was provided as a freely accessible web resource at the following URL: https://14-3-3-bindsite.streamlit.app/.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 9","pages":" 2602-2614"},"PeriodicalIF":6.2000,"publicationDate":"2025-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12360161/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00132c","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
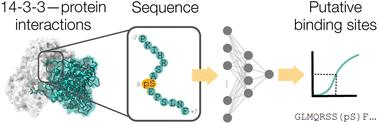
Protein–protein interactions are at the heart of biological processes. Understanding how proteins interact is key for deciphering their roles in health and disease, and for therapeutic interventions. However, identifying protein interaction sites, especially for intrinsically disordered proteins, is challenging. Here, we developed a deep learning framework to predict potential protein binding sites to 14-3-3 – a ‘central hub’ protein holding a key role in cellular signaling networks. After systematically testing multiple deep learning approaches to predict sequence binding to 14-3-3, we developed an ensemble model that achieved a 75% balanced accuracy on external sequences. Our approach was applied prospectively to identify putative binding sites across medically relevant proteins (ranging from highly structured to intrinsically disordered) for a total of approximately 300 sequences. The top eight predicted peptide sequences were experimentally validated in the wet-lab, and binding to 14-3-3 was confirmed for five out of eight sequences (Kd ranging from 1.6 ± 0.1 μM to 70 ± 5 μM). The relevance of our results was further confirmed by X-ray crystallography and molecular dynamics simulations. These sequences represent potential new binding sites within the 14-3-3 interactome (e.g., relating to Alzheimer's disease as the binding to tau is not the new part), and provide opportunities to investigate their functional relevance. Our results highlight the ability of deep learning to capture intricate patterns underlying protein–protein interactions, even for challenging cases like intrinsically disordered proteins. To further the understanding and targeting of 14-3-3/protein interactions, our model was provided as a freely accessible web resource at the following URL: https://14-3-3-bindsite.streamlit.app/.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


