Identification of Key Glycolysis-Related Genes in Osteoarthritis and Their Correlation with Immune Infiltration Using Bioinformatics Analysis and Machine Learning.
Yifang Zhu, Lin Deng, Junxiang Xia, Jing Yang, Dan Zhao, Min Li
{"title":"Identification of Key Glycolysis-Related Genes in Osteoarthritis and Their Correlation with Immune Infiltration Using Bioinformatics Analysis and Machine Learning.","authors":"Yifang Zhu, Lin Deng, Junxiang Xia, Jing Yang, Dan Zhao, Min Li","doi":"10.2147/OARRR.S541568","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Osteoarthritis (OA) is a degenerative disorder associated with glycolysis. However, the precise mechanisms remain unclear. This study aimed to identify glycolysis-associated biomarkers and elucidate how glycolysis-related genes interact with the synovial immune microenvironment in OA progression.</p><p><strong>Methods: </strong>Normal and OA synovial gene expression profile microarrays were obtained from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) were identified using limma package. Gene Ontology (GO) and KEGG enrichment analyses were conducted to explore biological functions. Weighted Gene Co-expression Network Analysis (WGCNA) was used to identify OA-associated genes, which were intersected with glycolysis genes from The Molecular Signatures Database (MSigDB) and DEGs to obtain key genes. Lasso regression and random forest models were employed to establish a risk model, and its predictive performance was evaluated using nomogram, Receiver Operating Characteristic (ROC) analysis, and Decision Curve Analysis (DCA). Gene Set Enrichment Analysis (GSEA) and Cibersort analysis were conducted to explore pathways and immune infiltration correlations.</p><p><strong>Results: </strong>A total of 239 OA-associated genes were identified through WGCNA. Six hub genes were obtained by intersecting with glycolysis genes and DEGs. Four key glycolytic genes were selected by Lasso regression and random forest models. The nomogram showed that three genes (DDIT4, SLC16A7, SLC2A3) could predict OA risk accurately. The ROC analysis demonstrated an area under the curve (AUC) of 0.85, indicating good predictive performance. Distinct immune cell distribution patterns were observed in OA groups. Interaction networks were constructed for the key genes with related miRNAs, transcription factors (TFs), and small molecule drugs.</p><p><strong>Conclusion: </strong>This study identified three key glycolysis-related genes (DDIT4, SLC16A7, SLC2A3) in OA, revealing their potential roles in disease progression and immune infiltration. These findings may provide new insights into the pathogenesis and therapeutic targets for OA, based on the identified genes and their interactions with the immune microenvironment.</p>","PeriodicalId":45545,"journal":{"name":"Open Access Rheumatology-Research and Reviews","volume":"17 ","pages":"157-171"},"PeriodicalIF":1.7000,"publicationDate":"2025-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12366638/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Open Access Rheumatology-Research and Reviews","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/OARRR.S541568","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"RHEUMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: Osteoarthritis (OA) is a degenerative disorder associated with glycolysis. However, the precise mechanisms remain unclear. This study aimed to identify glycolysis-associated biomarkers and elucidate how glycolysis-related genes interact with the synovial immune microenvironment in OA progression.
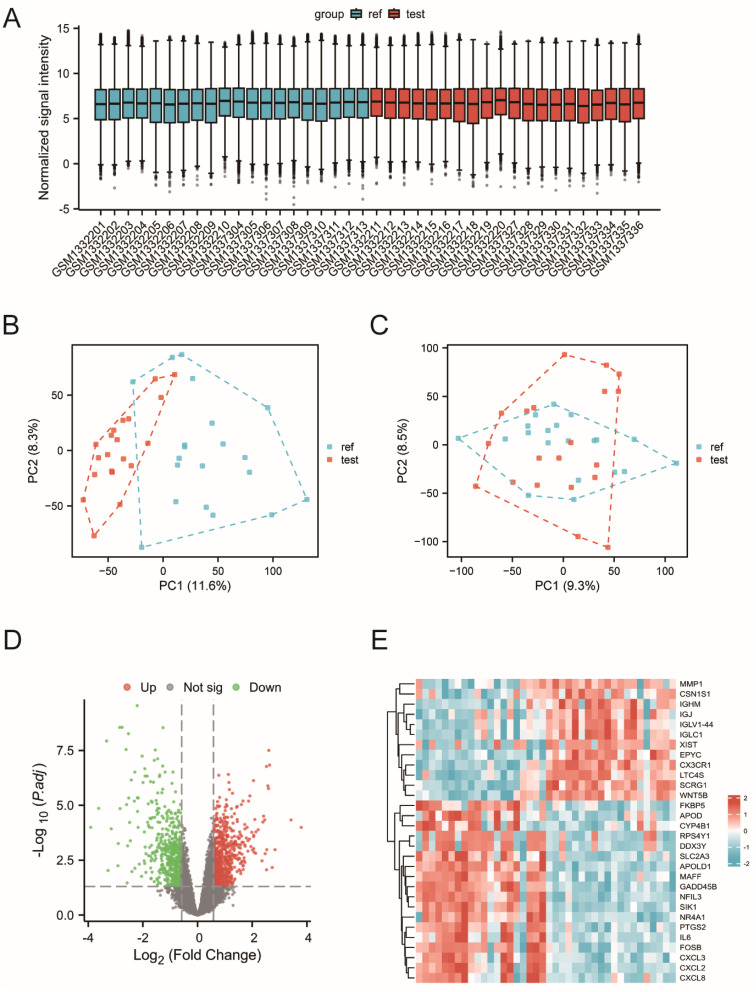
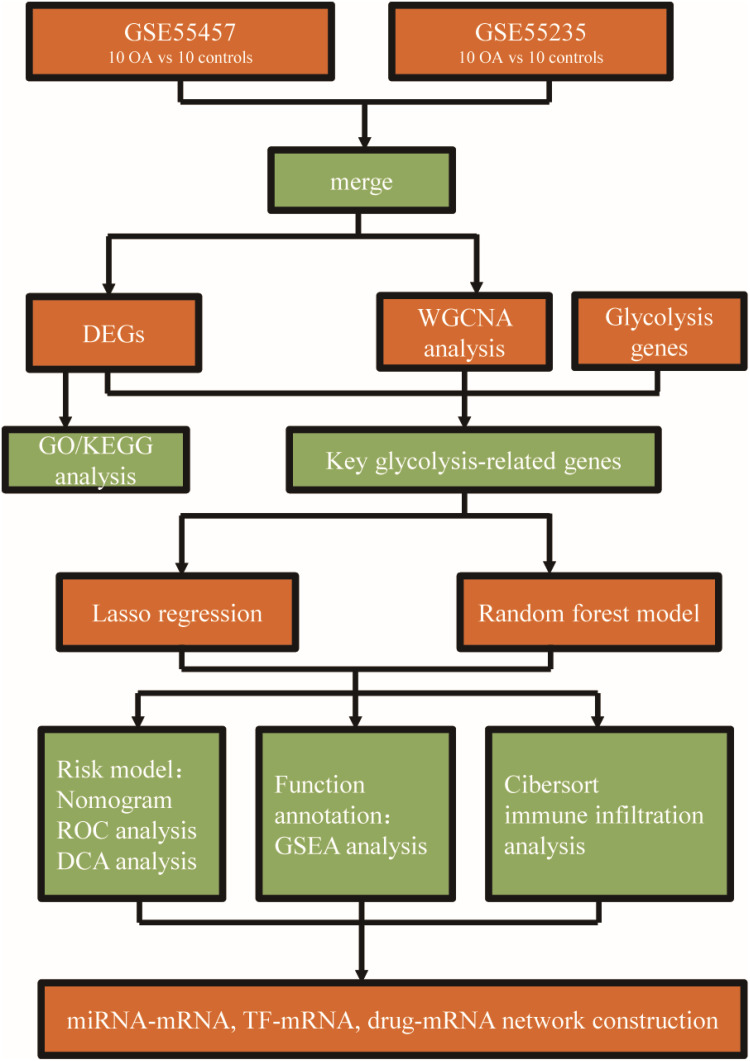
Methods: Normal and OA synovial gene expression profile microarrays were obtained from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) were identified using limma package. Gene Ontology (GO) and KEGG enrichment analyses were conducted to explore biological functions. Weighted Gene Co-expression Network Analysis (WGCNA) was used to identify OA-associated genes, which were intersected with glycolysis genes from The Molecular Signatures Database (MSigDB) and DEGs to obtain key genes. Lasso regression and random forest models were employed to establish a risk model, and its predictive performance was evaluated using nomogram, Receiver Operating Characteristic (ROC) analysis, and Decision Curve Analysis (DCA). Gene Set Enrichment Analysis (GSEA) and Cibersort analysis were conducted to explore pathways and immune infiltration correlations.
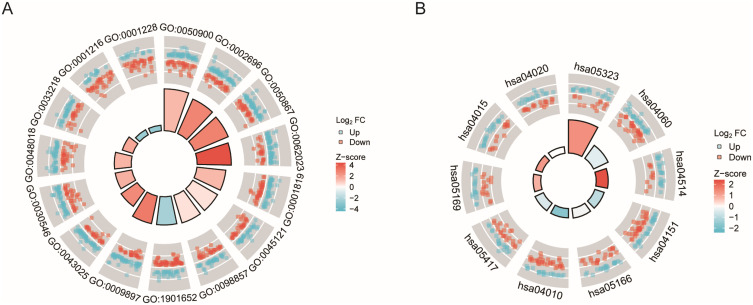
Results: A total of 239 OA-associated genes were identified through WGCNA. Six hub genes were obtained by intersecting with glycolysis genes and DEGs. Four key glycolytic genes were selected by Lasso regression and random forest models. The nomogram showed that three genes (DDIT4, SLC16A7, SLC2A3) could predict OA risk accurately. The ROC analysis demonstrated an area under the curve (AUC) of 0.85, indicating good predictive performance. Distinct immune cell distribution patterns were observed in OA groups. Interaction networks were constructed for the key genes with related miRNAs, transcription factors (TFs), and small molecule drugs.
Conclusion: This study identified three key glycolysis-related genes (DDIT4, SLC16A7, SLC2A3) in OA, revealing their potential roles in disease progression and immune infiltration. These findings may provide new insights into the pathogenesis and therapeutic targets for OA, based on the identified genes and their interactions with the immune microenvironment.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


