Said A Al-Busafi, Juland N Al Julandani, Zakariya Alismaeili, Juhaina J Al Raisi
{"title":"Wilson's Disease in Oman: A National Cohort Study of Clinical Spectrum, Diagnostic Delay, and Long-Term Outcomes.","authors":"Said A Al-Busafi, Juland N Al Julandani, Zakariya Alismaeili, Juhaina J Al Raisi","doi":"10.3390/clinpract15080144","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background/Objectives</b>: Wilson's disease (WD) is a rare autosomal recessive disorder of copper metabolism that results in hepatic, neurological, and psychiatric manifestations. Despite being described globally, data from the Middle East remains limited. This study presents the first comprehensive national cohort analysis of WD in Oman, examining clinical features, diagnostic challenges, treatment patterns, and long-term outcomes. <b>Methods</b>: A retrospective cohort study was conducted on 36 Omani patients diagnosed with WD between 2013 and 2020 at Sultan Qaboos University Hospital using AASLD diagnostic criteria. Clinical presentation, biochemical parameters, treatment regimens, and progression-free survival were analyzed. <b>Results</b>: The median age at diagnosis was 14.5 years, with a slight female predominance (55.6%). Clinical presentation varied: 25% had hepatic symptoms, 22.2% had mixed hepatic-neurological features, and 16.7% presented with neurological symptoms alone. Asymptomatic cases identified via family screening accounted for 33.3%. Diagnostic delays were most pronounced among patients presenting with neurological symptoms. A positive family history was reported in 88.9% of cases, suggesting strong familial clustering despite a low rate of consanguinity (5.6%). Regional distribution was concentrated in Ash Sharqiyah North and Muscat. Chelation therapy with trientine or penicillamine, often combined with zinc, was the mainstay of treatment. Treatment adherence was significantly associated with improved progression-free survival (<i>p</i> = 0.012). <b>Conclusions</b>: WD in Oman is marked by heterogeneous presentations, frequent diagnostic delays, and strong familial clustering. Early detection through cascade screening and sustained treatment adherence are critical for favorable outcomes. These findings support the need for national screening policies and structured long-term care models for WD in the region.</p>","PeriodicalId":45306,"journal":{"name":"Clinics and Practice","volume":"15 8","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2025-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12384659/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinics and Practice","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/clinpract15080144","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
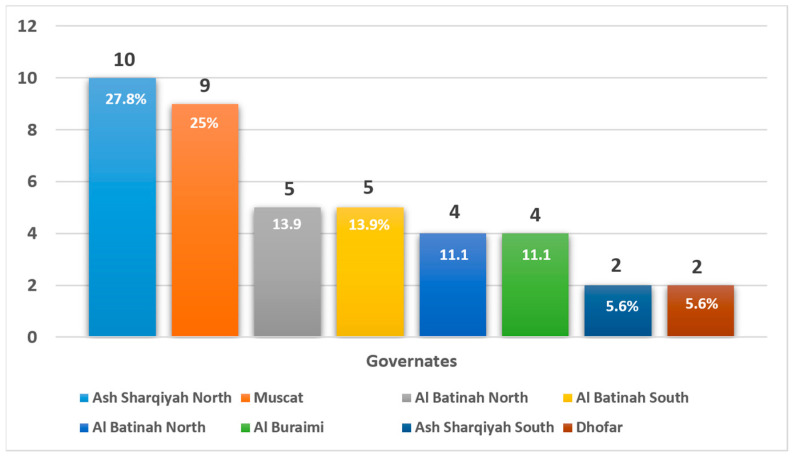
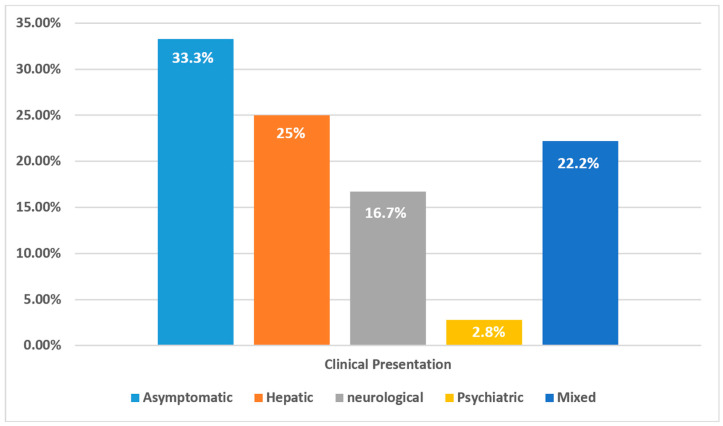
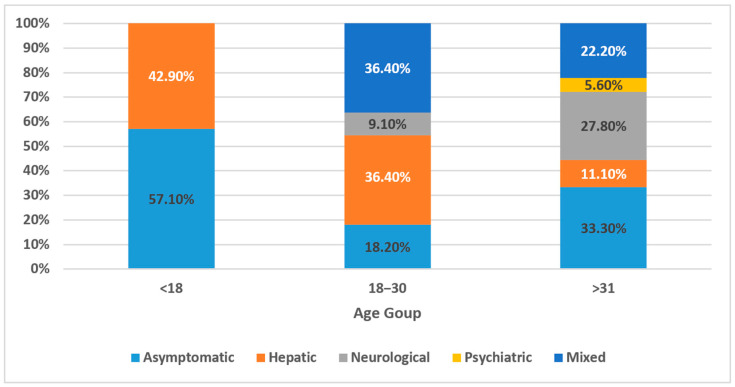
Background/Objectives: Wilson's disease (WD) is a rare autosomal recessive disorder of copper metabolism that results in hepatic, neurological, and psychiatric manifestations. Despite being described globally, data from the Middle East remains limited. This study presents the first comprehensive national cohort analysis of WD in Oman, examining clinical features, diagnostic challenges, treatment patterns, and long-term outcomes. Methods: A retrospective cohort study was conducted on 36 Omani patients diagnosed with WD between 2013 and 2020 at Sultan Qaboos University Hospital using AASLD diagnostic criteria. Clinical presentation, biochemical parameters, treatment regimens, and progression-free survival were analyzed. Results: The median age at diagnosis was 14.5 years, with a slight female predominance (55.6%). Clinical presentation varied: 25% had hepatic symptoms, 22.2% had mixed hepatic-neurological features, and 16.7% presented with neurological symptoms alone. Asymptomatic cases identified via family screening accounted for 33.3%. Diagnostic delays were most pronounced among patients presenting with neurological symptoms. A positive family history was reported in 88.9% of cases, suggesting strong familial clustering despite a low rate of consanguinity (5.6%). Regional distribution was concentrated in Ash Sharqiyah North and Muscat. Chelation therapy with trientine or penicillamine, often combined with zinc, was the mainstay of treatment. Treatment adherence was significantly associated with improved progression-free survival (p = 0.012). Conclusions: WD in Oman is marked by heterogeneous presentations, frequent diagnostic delays, and strong familial clustering. Early detection through cascade screening and sustained treatment adherence are critical for favorable outcomes. These findings support the need for national screening policies and structured long-term care models for WD in the region.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


