Creating a consensus genome assembly of Myxococcus xanthus DZ2 by resolving discrepancies between two complete genomes of the same strain and uncovering the Mx-alpha prophage region diversity across phylum Myxococcota.
{"title":"Creating a consensus genome assembly of <i>Myxococcus xanthus</i> DZ2 by resolving discrepancies between two complete genomes of the same strain and uncovering the Mx-alpha prophage region diversity across phylum Myxococcota.","authors":"Utkarsha Mahanta, Gaurav Sharma","doi":"10.1093/nargab/lqaf112","DOIUrl":null,"url":null,"abstract":"<p><p><i>Myxococcus xanthus</i> DZ2, a model myxobacterium, has three reported genome assemblies, including two recent complete assemblies (MxDZ2_Tam and MxDZ2_Nan) from the same culture stock. These assemblies misreported their circular nature and differed by 6.4 kb, raising questions about their accuracy. After removing duplicate ends, aligning genomes to the origin of replication, and circularization, this computational analysis revealed a minimal 32 bp difference, with MxDZ2_Tam being slightly larger. Forty sequence variations including 38 indels and two substitutions, were impacting 18 coding genes via frameshift mutations. Although PacBio-HiFi technology boasts a low error rate, it remains higher than the 454-platform used for the earlier MxDZ2_Kirby draft assembly. Therefore, using MxDZ2_Kirby as a reference, we constructed a \"truly circular\" genome for <i>M. xanthus</i> DZ2. Additionally, analysis of Mx-alpha regions, involved in antagonism via the toxin gene <i>sitA</i>, across 61 myxobacterial genomes identified their presence in five taxonomically polyphyletic species, potentially influencing their physiology, development, and ecological interactions beyond predation. Only <i>M. xanthus</i> DZ2 and DZF1 contained all three Mx-alpha regions, whereas <i>M. xanthus</i> DK1622 has only one. Overall, this study underscores the need for meticulous validation of sequencing-based genome assemblies and their variations and provides novel insights into Mx-alpha regions as potential adaptive elements in myxobacteria.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"7 3","pages":"lqaf112"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12390754/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqaf112","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
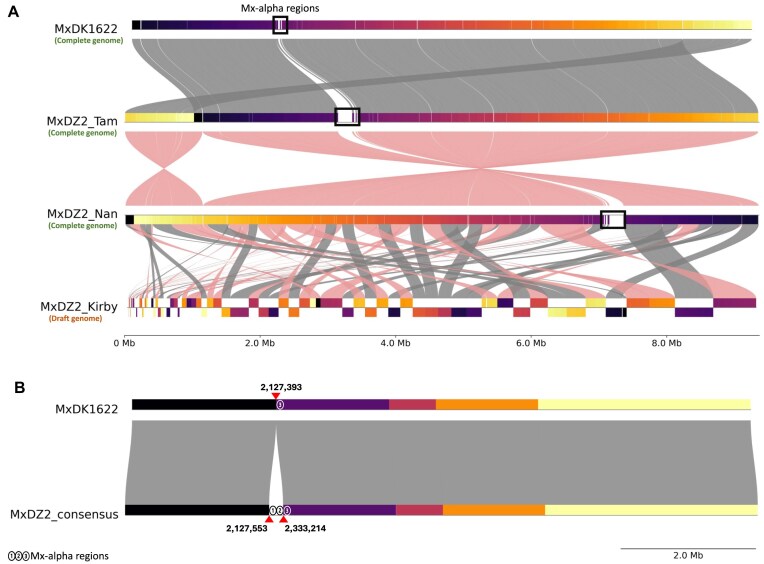
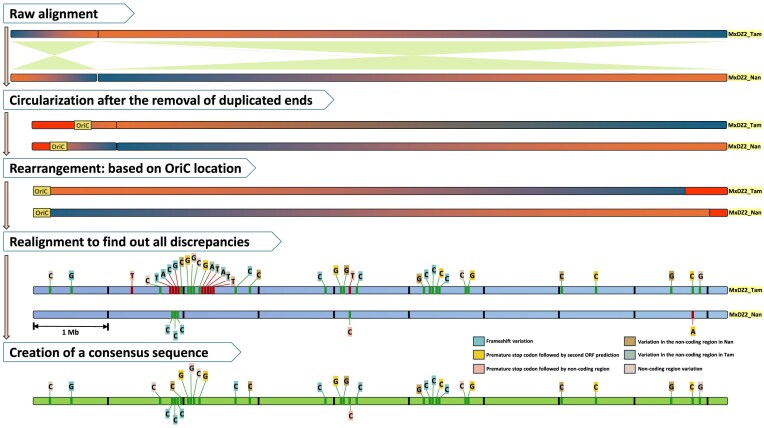
Myxococcus xanthus DZ2, a model myxobacterium, has three reported genome assemblies, including two recent complete assemblies (MxDZ2_Tam and MxDZ2_Nan) from the same culture stock. These assemblies misreported their circular nature and differed by 6.4 kb, raising questions about their accuracy. After removing duplicate ends, aligning genomes to the origin of replication, and circularization, this computational analysis revealed a minimal 32 bp difference, with MxDZ2_Tam being slightly larger. Forty sequence variations including 38 indels and two substitutions, were impacting 18 coding genes via frameshift mutations. Although PacBio-HiFi technology boasts a low error rate, it remains higher than the 454-platform used for the earlier MxDZ2_Kirby draft assembly. Therefore, using MxDZ2_Kirby as a reference, we constructed a "truly circular" genome for M. xanthus DZ2. Additionally, analysis of Mx-alpha regions, involved in antagonism via the toxin gene sitA, across 61 myxobacterial genomes identified their presence in five taxonomically polyphyletic species, potentially influencing their physiology, development, and ecological interactions beyond predation. Only M. xanthus DZ2 and DZF1 contained all three Mx-alpha regions, whereas M. xanthus DK1622 has only one. Overall, this study underscores the need for meticulous validation of sequencing-based genome assemblies and their variations and provides novel insights into Mx-alpha regions as potential adaptive elements in myxobacteria.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


