{"title":"Beyond rigid docking: deep learning approaches for fully flexible protein-ligand interactions.","authors":"John Lee, Canh Hao Nguyen, Hiroshi Mamitsuka","doi":"10.1093/bib/bbaf454","DOIUrl":null,"url":null,"abstract":"<p><p>Sparked by AlphaFold2's groundbreaking success in protein structure prediction, recent years have seen a surge of interest in developing deep learning (DL) models for molecular docking. Molecular docking is a computational approach for predicting how proteins interact with small molecules known as ligands. It has become an essential tool in drug discovery, enabling structure-based virtual screening (VS) methods to efficiently explore vast libraries of drug-like molecules and identify potential therapeutic candidates. However, traditional docking methods primarily rely on search-and-score algorithms, which are computationally demanding. To be viable for VS applications, these methods often sacrifice accuracy for speed by simplifying their search algorithms and scoring functions. Recent advancements in DL have transformed molecular docking, offering accuracy that rivals-or even surpasses-traditional approaches while significantly reducing computational costs. Despite these advancements, DL-based molecular docking still faces major challenges. DL models often struggle to generalize beyond their training data and frequently mispredict key molecular properties, such as stereochemistry, bond lengths, and steric interactions, leading to physically unrealistic predictions. To overcome these limitations, a new generation of models is using DL to incorporate protein flexibility into docking predictions, aiming to more accurately capture the dynamic nature of biomolecular interactions-a long-standing challenge for traditional methods. This review explores how DL has reshaped molecular docking, examines its current shortcomings, and highlights emerging solutions. Finally, we discuss future opportunities to further bridge the gap between computational predictions and real-world molecular interactions.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"26 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406700/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbaf454","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
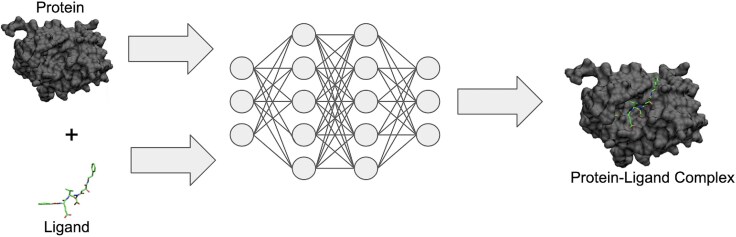
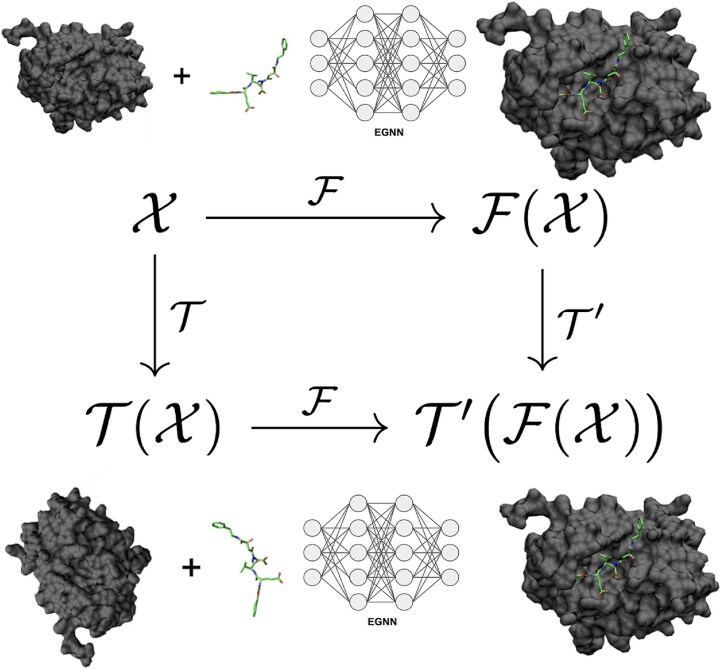
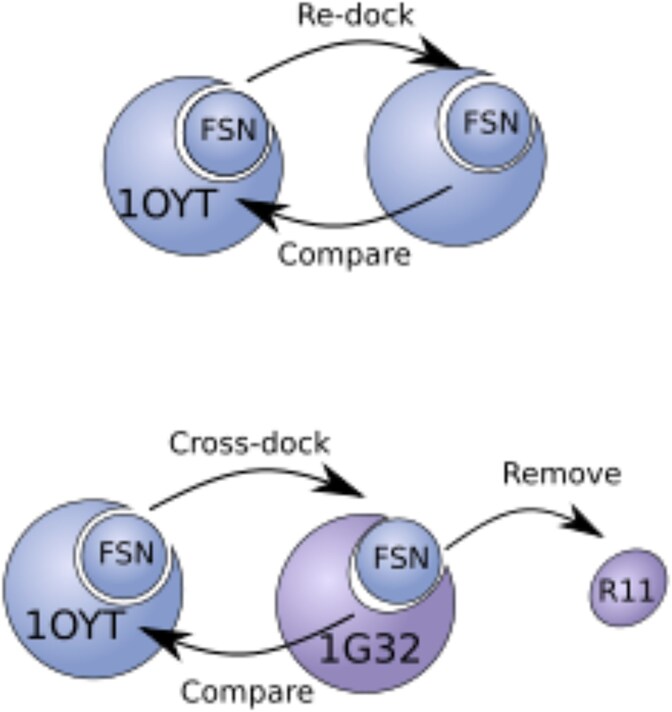
Sparked by AlphaFold2's groundbreaking success in protein structure prediction, recent years have seen a surge of interest in developing deep learning (DL) models for molecular docking. Molecular docking is a computational approach for predicting how proteins interact with small molecules known as ligands. It has become an essential tool in drug discovery, enabling structure-based virtual screening (VS) methods to efficiently explore vast libraries of drug-like molecules and identify potential therapeutic candidates. However, traditional docking methods primarily rely on search-and-score algorithms, which are computationally demanding. To be viable for VS applications, these methods often sacrifice accuracy for speed by simplifying their search algorithms and scoring functions. Recent advancements in DL have transformed molecular docking, offering accuracy that rivals-or even surpasses-traditional approaches while significantly reducing computational costs. Despite these advancements, DL-based molecular docking still faces major challenges. DL models often struggle to generalize beyond their training data and frequently mispredict key molecular properties, such as stereochemistry, bond lengths, and steric interactions, leading to physically unrealistic predictions. To overcome these limitations, a new generation of models is using DL to incorporate protein flexibility into docking predictions, aiming to more accurately capture the dynamic nature of biomolecular interactions-a long-standing challenge for traditional methods. This review explores how DL has reshaped molecular docking, examines its current shortcomings, and highlights emerging solutions. Finally, we discuss future opportunities to further bridge the gap between computational predictions and real-world molecular interactions.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


